- Made available online as an Accepted Preprint 24 February 2009
- Accepted Preprint first posted online on 24 February 2009
Bone loss in inflammatory disorders
- R Hardy and
- M S Cooper
- School of Clinical and Experimental Medicine, Institute of Biomedical Research, University of Birmingham, Birmingham B15 2TT, UK
- (Correspondence should be addressed to M S Cooper who is now at Endocrinology, School of Clinical and Experimental Medicine, Queen Elizabeth Hospital, University of Birmingham, Edgbaston, Birmingham B15 2TH, UK; Email: m.s.cooper{at}bham.ac.uk)
Abstract
Chronic inflammatory diseases of almost any cause are associated with bone loss. Bone loss is due to direct effects of inflammation, poor nutrition, reduced lean body mass, immobility and the effects of treatments, especially glucocorticoids. These mechanisms are complex and interrelated but are ultimately mediated through effects on the bone remodelling cycle. Inflammatory disease can increase bone resorption, decrease bone formation but most commonly impacts on both of these processes resulting in an uncoupling of bone formation from resorption in favour of excess resorption. This review will illustrate these interactions between inflammation and bone metabolism and discuss how these are, and might be, manipulated as therapies for inflammation related bone loss.
Introduction
Chronic inflammatory diseases are frequently associated with systemic bone loss. The mechanisms underlying this bone loss are complex and interrelated. These mechanisms appear, however, to be ultimately mediated through effects on the bone remodelling cycle. Inflammatory disease can increase bone resorption, decrease bone formation but most commonly impacts on both of these processes. This review will illustrate these interactions between inflammation and bone metabolism and discuss how these are, and might be, manipulated as therapies for inflammation related bone loss.
Relationship between inflammation and bone disease
A relationship between inflammation and bone disease has been established in a variety of clinical settings and animal models of inflammatory disease (Spector et al. 1993, Gough et al. 1994, Bernstein et al. 2000, Schoon et al. 2000, Bultink et al. 2005). It is clear that the nature of the inflammatory disease can impact on the extent and type of bone disease and that even low level sub-clinical inflammation has been reported to impact on bone remodelling and increase fracture risk (Schett et al. 2006). Although inflammation can affect almost any organ of the body most interest has been focussed on the commonest chronic inflammatory diseases seen clinically. These are inflammatory joint disease (best exemplified by rheumatoid arthritis), inflammatory bowel disease (e.g. Crohn's disease or ulcerative colitis), coeliac disease, lung inflammation (asthma, chronic obstructive pulmonary disease, alveolitis), renal disease (nephritis, vasculitis) and disease affecting nerve and muscle (myositis, inflammatory neuropathy). These diseases share many mechanisms in common by which bone can be lost but each has features distinct from the others. Treatments for these conditions are themselves implicated in bone loss and these will also vary with type of disease.
Potential mediators of bone loss
Various mechanisms have been proposed by which bone is lost during inflammation. Systemic inflammation can directly affect bone resorption and/or bone formation leading usually to bone loss, but rarely, bone gain. Bone is also very sensitive to nutritional state. Most chronic inflammatory diseases are associated with a catabolic state that favours the loss of lean mass and this reduces bone formation. Diseases that affect the gastrointestinal tract will have an additional impact on the intake of nutrients and calories. A commonly used treatment for many inflammatory diseases is glucocorticoids. These drugs frequently have a major adverse effect on bone that is difficult to separate from the effects of inflammation itself. Inflammation also has an impact on the control of reproductive hormones leading frequently to hypogonadism in both men and women. Inflammation also impacts on the secretion and/or action of PTH which can increase bone resorption. An additional problem is that chronic inflammatory diseases are often associated with either immobility or reduced exercise tolerance. This reduction in activity can itself lead to bone loss through reduced mechanical stimulation of bone. A unifying theme among these mechanisms is that they all impact on the bone remodelling cycle.
The bone remodelling cycle
The bone remodelling cycle refers to the coordinated interaction between osteoclasts and osteoblasts to initially remove an area of bone (a process carried out by osteoclasts) and then replace the lost bone with new matrix (a process carried out by osteoblasts) which subsequently mineralises. The remodelling cycle is essential for the repair of damage at a microscopic level within bone. Bone remodelling occurs normally in all individuals and in adults about 25% of trabecular and 3% of cortical bone is replaced through remodelling each year (Manolagas & Jilka 1995). An imbalance in the amount of bone removed relative to that replaced during each cycle in favour of excessive bone resorption will lead to a decline in the total amount of bone and will increase the risk of fractures. In most situations, the amount of bone formed closely matches the amount lost but with ageing there is a small negative balance leading to the age-related decline in bone density.
The remodelling cycle is controlled by a variety of endocrine and immunological mechanisms (Zhao et al. 2006, Goldring & Goldring 2007, Matsuo & Irie 2008, Sims & Gooi 2008). The discovery of the osteoprotegerin (OPG)/receptor activator of nuclear factor-κB ligand (RANKL) system has given insight into a major component of the remodelling cycle. RANKL is expressed on the surface of osteoblasts and its expression increases in response to a variety of pro-resorptive signals such as proinflammatory cytokines, glucocorticoids, oestrogen deficiency and PTH excess (Hofbauer et al. 2000). RANKL binds to the RANK receptor which is expressed on osteoclasts and their precursors. RANKL is a critical stimulator of the differentiation and activity of osteoclasts and thus the promotion of bone resorption. OPG is a decoy receptor for RANKL that is secreted by osteoblasts (and to a lesser extent other stromal derived cells) and acts to reduce bone resorption. The importance of OPG and RANKL are highlighted in various mouse models, including mice lacking RANK or RANKL, or overexpressing OPG (Simonet et al. 1997, Dougall et al. 1999, Kong et al. 1999b). These mice lack active osteoclasts resulting in osteopetrosis with virtually no bone remodelling. Conversely, overexpression of RANK or RANKL or knockout of OPG leads to severe osteoporosis due to excessive osteoclast activity.
The OPG/RANKL system accounts only for signalling of osteoblasts to osteoclasts. Other signalling pathways are likely to exist where osteoclasts can regulate bone formation. Recently, bidirectional signals that can ‘couple’ formation to resorption such as the Ephs and Ephrins have been identified in osteoblasts and osteoclasts (Zhao et al. 2006). There are likely to be several physiological regulators of communication between osteoblasts and osteoclasts that attempt to maintain the balance between bone resorption and formation and these mechanisms will need to be bypassed or overcome during inflammation to induce bone loss. An overview of how inflammatory disease affects the bone remodelling cycle is shown in Fig. 1. The impact of inflammation related mechanisms of bone loss on the bone remodelling cycle will be discussed along with how inflammation within particular tissues interacts with bone remodelling.

Illustration of how chronic inflammatory disease impacts on bone formation and resorption. A stimulatory effect is indicated by + and an inhibitory effect by −.
Systemic inflammation
A major advance in our understanding of inflammation related bone loss was the observation that proinflammatory cytokines could stimulate osteoclastogenesis (Thomson et al. 1987). This effect is best exemplified by the effect of tumour necrosis factor-α (TNF-α) on expression of RANKL by osteoblasts. As discussed above, RANKL is able to induce osteoclast differentiation and stimulates bone resorbing activity. TNF-α has an additional direct pro-resorbative action on osteoclasts in vitro (Kobayashi et al. 2000, Kim et al. 2005). This effect is probably of limited physiological importance in vivo since TNF-α is very poor in inducing osteoclastogenesis in RANK deficient mice (Li et al. 2000). TNF-α and interleukin-1 (IL-1) can, however, synergise with RANKL to directly potentiate bone resorption by osteoclasts. Although under normal circumstances, RANKL is derived from osteoblasts during inflammation a range of inflammatory cells can also generate RANKL. These cells include lymphocytes and fibroblasts e.g. those found in the inflamed synovium (Kong et al. 1999a, Gravallese et al. 2000, Kotake et al. 2001). RANKL can exert its effects in a membrane bound or soluble form but the membrane bound form appears more effective at inducing osteoclastogenesis (Nakashima et al. 2000). The expression of RANKL on non-osteoblastic cells or the release of RANKL can thus lead to a direct osteoclastogenic signal independent of osteoblasts. Since the discovery of the OPG/RANKL system a wide variety of other cytokines have been found to impact on this system or to directly affect osteoclastogenesis. Some cytokines have stimulatory effects on osteoclastogenesis (e.g. TNF-α, IL-1β, IL-6, IL-11 and IL-17) whereas others have predominantly inhibitory effects (e.g. interferon (IFN)-γ, IL-4 and transforming growth factor-β; Lorenzo et al. 2008). The balance of these cytokines is likely to differ between disease states potentially accounting for differences in predisposition to bone loss. Recently, it has been proposed that T lymphocytes from the classical Th1 and Th2 lymphocyte subsets probably secrete a pattern of cytokines that is inhibitory to osteoclastogenesis and yet in many inflammatory diseases, there is a T-cell mediated increase in osteoclastogenesis (Sato et al. 2006). The explanation for this appears to be that lymphocytes from the recently identified Th17 subset, named after the ability of these lymphocytes to secrete IL-17, have a particularly osteoclastogenic cytokine profile. This lymphocyte subset is particularly prominent in inflammatory arthritis and thus could explain the predisposition to local osteoclast formation and bone destruction in this condition (Lundy et al. 2007). These cells are also implicated in inflammatory bowel disease and asthma (Tesmer et al. 2008).
An increase in bone resorption should secondarily result in a stimulation of bone formation due to the normally tight coupling of bone resorption and formation. However, in the majority of situations where chronic inflammation persist bone formation is suppressed or remains inappropriately normal relative to the degree of resorption. The explanation for this is presently unclear. It is possible that proinflammatory cytokines could additionally directly suppress bone formation. There is evidence that TNF-α can inhibit the differentiation of osteoblasts (Gilbert et al. 2000). It is also possible that inflammation could directly interrupt the signalling mechanisms that couple formation to resorption. This is difficult to assess because the mechanisms that mediate this limb of coupling remain poorly understood.
An elegant explanation for the uncoupling of bone formation from resorption has emerged from animal models of inflammatory arthritis and implicates the wnt signalling pathway, and the wnt antagonist dickkopf-1 (DKK1) in particular, in this effect (Diarra et al. 2007). Recent studies suggest that the canonical wnt signalling pathway is central in bone development, regulating differentiation of mesenchymal precursor cells into mature osteoblasts, as well as playing a central role in the normal development of the skeleton in the embryo (Johnson et al. 2004, Westendorf et al. 2004, Hu et al. 2005). Patients with mutations of the low density lipoprotein receptor-related protein 5 (LRP5) wnt coreceptor that cause constitutive receptor activation have a grossly increased bone density with improved strength (referred to as the high bone mass phenotype). By contrast, mutations of LRP5 that lead to inactivation of the wnt signalling pathway are associated with severe osteoporosis as part of the osteoporosis-pseudoglioma syndrome (Gong et al. 2001, Boyden et al. 2002, Little et al. 2002). Wnts act by binding to the frizzled/LRP5 receptor complex leading to the inactivation of glycogen synthase kinase-3b, and inhibition of phosphorylation and degradation of β-catenin (Fig. 2; Wodarz & Nusse 1998, Bejsovec 2000). This results in the accumulation of β-catenin, which translocates into the cell nucleus and forms a complex with transcription factors from the T-cell factor and lymphoid enhancer families. Inhibition of wnt signalling by soluble wnt antagonists such as DKK1 blocks this process. The importance of DKK1 in normal bone remodelling is demonstrated by models such as the DKK1 knockout mouse, which displays an increased bone mass, while myeloma cells with aberrant DKK1 expression are associated with purely lytic lesions with little evidence of bone formation (Tian et al. 2003, Morvan et al. 2006). The secretion of DKK1 by synovial fibroblasts was found to be increased by TNF-α and circulating levels of DKK1 were elevated in patients with rheumatoid arthritis (Diarra et al. 2007). DKK1 secreted from the synovium could inhibit local bone formation through a suppressive effect on osteoblast maturation. This suppression of bone formation was associated with increased resorption. This increased resorption was thought to be due to effects of DKK1 on the OPG-RANKL system. Inhibition of DKK1 by a neutralising antibody led to an elevation in systemic OPG levels and blockage of local OPG production by the injection of siRNA for OPG into the joint led to a re-emergence of osteoclasts. Most importantly, administration of an antibody to DKK1 could prevent bone erosions and reverse this block on osteoblast formation. The DKK1 antibody treatment also resulted in a paradoxical excess of bone formation during inflammation as evidenced by the development of new osteophytes. As such the wnt pathway and DKK1 in particular, has been proposed to be the central regulator of bone remodelling in inflammatory arthritis (summarised in Fig. 3). Further examination of the whole wnt pathway (which consists of a variety of agonists, antagonists, receptors and coreceptors) will hopefully shed more light on the regulation of bone remodelling and the coupling of formation and resorption in inflammatory arthritis. The role of the wnt signalling pathway in other inflammatory conditions (where there is not the capacity of synovial fibroblasts to impact on bone) will need further examination (Herman et al. 2008) although wnt signalling is implicated in immobility related and glucocorticoid induced bone loss as discussed below.

The canonical Wnt pathway: the wnt pathway increases β-catenin availability. In the absence of Wnt signalling, a destruction complex containing axin, glycogen synthase kinase-3b (GSK-3b), adenomatous polypsis coli (APC) and casein kinase 1 (CK1) targets β-catenin for proteosomal degradation. Wnt signalling is initiated by binding of Wnts to the frizzled (Fz) and LRP5/6 co-receptor complex. Activation causes recruitment of GSK-3β binding protein (GBP), which interferes with β-catenin degradation. β-Catenin accumulates within the cytosol and translocates into the nuclues where it binds to TCF/LEF transcription factors and initiates transcription of Wnt target genes. Several secreted factors inhibit Wnt signalling through binding to Wnts (e.g. secreted Fz related proteins (SFrp) and Wif1) or the LRP5/6 receptor (e.g. DKK1 and sclerostin).
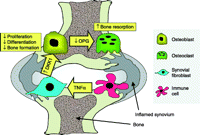
Schematic illustration of the possible role of DKK1 in the bone remodelling imbalance in inflammatory joint disease. Production of DKK1 in response to TNF-α production by inflammatory cells is proposed to inhibit bone formation but increase bone resorption by osteoclasts through a suppression of OPG production by osteoblasts.
An additional mechanism by which inflammation could uncouple bone formation from resorption is through alteration of glucocorticoid signalling. The effects of glucocorticoids are discussed below in the context of therapeutic glucocorticoids but inflammation is a potent modifier of local glucocorticoid action in bone cells. It is now clear that the levels of active glucocorticoids present within the circulation can differ from that in the tissues due to tissue metabolism of glucocorticoids by intracellular enzymes (Tomlinson et al. 2004). The main enzymes responsible for tissue metabolism of glucocorticoids are the 11β-hydroxysteroid dehydrogenases (11β-HSDs). The 11β-HSD1 enzyme converts inactive glucocorticoids such as cortisone and prednisone to their active counterparts cortisol and prednisolone. 11β-HSD1 is expressed in osteoblasts and can increase local glucocorticoid action in these cells. Overexpression of the enzyme in osteoblasts decreases proliferation and the synthesis of bone specific proteins such as osteocalcin when cells are exposed to inactive glucocorticoids (Cooper et al. 2000, Rabbitt et al. 2002). We have previously shown that proinflammatory cytokines such as TNF-α or IL-1β can potently induce the expression and activity of this enzyme in osteoblasts (Cooper et al. 2001). This effect was functionally important as evidenced by the induction of glucocorticoid target genes. Thus, during inflammation, osteoblasts within bone that are exposed to proinflammatory cytokines are likely to also be exposed to high doses of autocrine generated cortisol (Cooper et al. 2001, Canalis & Delany 2002). This is potentially a major mechanism by which osteoblasts and osteoclasts are uncoupled. A high glucocorticoid level in osteoblasts will decrease bone formation through direct effects on osteoblasts (Cooper 2004) but is also able to maintain an osteoclastogenic signal due to upregulation of RANKL and downregulation of OPG in osteoblast precursors (Hofbauer et al. 1999). The relative importance of locally generated glucocorticoids, and whether there is any interaction of this with other proposed mechanisms of uncoupling, such as DKK1 induction remains to be clarified.
Nutrition
Chronic inflammation is frequently complicated by poor nutrition. This is partly due to inflammation directly inducing a highly catabolic state. This ‘cachexia’ is multifactorial and is at least partly mediated through proinflammatory cytokines (Rall & Roubenoff 2004). It is predominantly manifested by a reduction in lean body mass. Chronic inflammation of the gastrointestinal tract can have an additional impact through reduced calorie intake and difficulty in absorbing specific nutrients important to bone that rely on complex absorptive mechanisms. The most obvious examples of the role of poor nutrition in inflammatory bowel disease are calcium and vitamin D malabsorption in coeliac disease (Corazza et al. 2005). Calcium and vitamin D are important in maintaining adequate mineralization of bone. Deficiency is associated with attempts by the body to overcome the problem by increasing PTH secretion. PTH receptors are expressed on osteoblasts and persistent PTH signalling results in an increase in RANKL expression on osteoblasts. This increased RANKL expression increases bone resorption through an increase in osteoclastogenesis. Independent from direct effects on the bone remodelling cycle, persistent deficiency in calcium or vitamin D intake can affect the mineralization of bone which can independently increase risk of fracture. A further mechanism by which vitamin D deficiency can be induced is via the immobilising effect of chronic illness. Vitamin D synthesis from skin, which is the major source of vitamin D, can only occur with exposure to the sun (and u.v. radiation in particular) which is frequently reduced in patients with chronic disease (Cooper & Gittoes 2008).
An additional complicating factor is the potential role of vitamin D in modulating the immune response, and thus an indirect role in inflammation associated bone loss. An anti-inflammatory role of vitamin D has been implicated in conditions as diverse as renal inflammation, rheumatoid arthritis and inflammatory bowel disease (Merlino et al. 2004, Froicu & Cantorna 2007, Zehnder et al. 2008). These studies have demonstrated a correlation of the degree of inflammation with low levels of vitamin D. It is now clear that some cells within the immune system have the capacity to generate the most active form of vitamin D, 1,25-dihydroxyvitamin D, from the circulating precursor 25-hydroxyvitamin D (Adams & Hewison 2008). This ability can influence the function of antigen-presenting cells and the capacity of macrophages to kill intracellular pathogens e.g. mycobacteria (Hewison et al. 2003, Liu et al. 2006). There are, however, no trials that demonstrate that supplementation with vitamin D is able to modulate inflammatory disease in a clinical setting.
Patients with inflammatory bowel disease (Crohn's disease or ulcerative colitis) frequently have very poor nutritional intake resulting in low total and lean body mass. This has been proposed to be at least as, if not more important, than the direct effects of inflammation (Burnham et al. 2007). The predominant bone remodelling abnormality in inflammatory bowel disease appears to be a reduction in bone formation with inappropriately maintained bone resorption (Sylvester et al. 2007). In other states characterised by poor nutritional availability such as anorexia nervosa a similar uncoupling of bone resorption from formation with suppressed formation is seen (Soyka et al. 1999). The mechanisms underlying this response to poor nutrition are not known. Even though these changes coincide with hypogonadism, oestrogen replacement fails to improve the poor bone formation. It is now clear that at least part of the remodelling of bone is governed by central inputs (Karsenty 2006). Factors such as leptin can signal in the central nervous system and have impacts on bone affected through the sympathetic nervous system. These centrally mediated signalling pathways are probably critical to the low bone formation seen during anorexia and low lean mass. Presently, there are no pharmacological approaches available to manipulate the central regulation of bone mass.
The bone loss that is seen in an acute flare of colitis appears to be very rapid but there is the potential for rapid return of bone density on successful treatment of the disease (Tobias et al. 2004). In a clinical setting, the question of how inflammation affects bone during colitis is frequently confounded by the use of glucocorticoids as the predominant anti-inflammatory agent.
Glucocorticoids
An excess of circulating glucocorticoid has a dramatic adverse effect on bone (Manolagas & Weinstein 1999, Cooper 2004, Canalis et al. 2007). The hallmark of glucocorticoid excess is disruption of bone remodelling with uncoupling of formation from resorption (Fig. 4). Glucocorticoids cause early and profound reduction in formation through direct effects on osteoblasts (Pearce et al. 1998, Cooper et al. 2003). Glucocorticoids decrease osteoblast proliferation and production of osteoblast specific proteins (e.g. osteocalcin, bone specific alkaline phosphatase, N-terminal propeptide of type I collagen), and increase apoptosis (Bellows et al. 1990, Weinstein et al. 1998, Eijken et al. 2006). Glucocorticoids interfere with a range of important signalling pathways in osteoblasts. These include IGF-1 and wnt signalling (Wang et al. 2008). Glucocorticoid excess also results in increased or inappropriate bone resorption despite the reduction in the formation. This effect is also thought to be via osteoblasts through a decrease in OPG and increase in RANKL expression (Hofbauer et al. 1999). With long term glucocorticoid, excess resorption falls possibly due to inhibition of differentiation of osteoclast precursors (Weinstein et al. 2002).
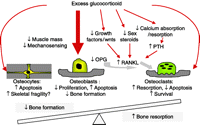
Illustration of the mechanisms by which excess glucocorticoids have an effect on bone. The net result is an uncoupling of bone formation and resorption in favour of bone loss.
The effects of glucocorticoids are often difficult to distinguish from those of the underlying inflammatory illness since high doses of glucocorticoids are rarely used in non-inflammatory settings. High circulating levels of glucocorticoids are seen in endogenous Cushing's syndrome e.g. caused by an adrenal adenoma secreting cortisol or a pituitary adenoma secreting ACTH (which then stimulates cortisol release). Bone loss is a common feature in this situation implying that glucocorticoids can induce bone disease in isolation (Chiodini et al. 1998, Fernandez-Rodriguez et al. 2008). Additionally, a trial of high dose glucocorticoids in men with antisperm antibodies demonstrated that bone formation markers were suppressed with inappropriately maintained levels of bone resorption markers (Pearce et al. 1998). This combination was associated with rapid and substantial systemic bone loss when assessed by bone densitometry. Despite these observations, it appears that the effects of therapeutic glucocorticoids are partly dependent on an interaction with the underlying illness (Leonard et al. 2004, Burnham et al. 2007). Glucocorticoids can suppress inflammation and help to resolve the underlying illness. This might help to protect against bone loss despite their adverse affects on bone (Kirwan 1995, Hansen et al. 1996). On the other hand, glucocorticoids have been reported to have much less of an adverse effect on bone in patients with non-inflammatory conditions than in the presence of inflammation. This relative sparing of bone density has best been illustrated in children with glucocorticoid sensitive nephrotic syndrome (Leonard et al. 2004). These patients require very high doses of therapeutic steroids for long periods of time and would thus be expected to have a substantial impairment of bone density. However, these patients appear to have well preserved bone density. This is in dramatic contrast to children taking glucocorticoids for inflammatory disorders (Lien et al. 2003). The relative lack of a negative effect in the absence of inflammation was associated with an increase in obesity and this increase in body mass index might have a protective effect through increased mechanical stimulation due to the increased weight. In inflammatory disease, this beneficial effect was not seen since the amount of lean mass was reduced. The relationship between glucocorticoid damage and the degree of inflammation may depend on the skeletal site examined with differences between trabecular and cortical bone reported (Wetzsteon et al. 2009). Trabecular bone appears to be sensitive to the effects of glucocorticoids in all situations whereas cortical bone was relatively spared, or even increased in density, in non-inflammatory disease.
Another way in which inflammation might modify the effect of therapeutic glucocorticoids in inflammatory disease is through modulation of the 11β-HSD1 system that was described earlier. 11β-HSD1 activity within bone appears to be the major determinant of individual sensitivity to the effects of therapeutic glucocorticoids in healthy individuals without inflammatory disease (Cooper et al. 2003). However, as discussed above, 11β-HSD1 is induced by inflammatory cytokines. This induction appears to be tissue dependent and the extent of induction can vary between sites even in the same tissue (Tomlinson et al. 2001, Hardy et al. 2006). What is clear is that tissues that express 11β-HSD1 during inflammation will have a higher relative exposure to glucocorticoids when using drugs that are substrates for this enzyme. Inflammation, especially if in proximity to bone, is thus potentially able to sensitise bone to therapeutic glucocorticoids (Cooper et al. 2001). An additional way in which 11β-HSD1 activity could influence bone is through paracrine effects due to glucocorticoid activation in non- bone tissue. We have recently reported that synovium in patients with inflammatory arthritis has substantial glucocorticoid activating activity leading to high local levels of glucocorticoids within the inflamed joint (Hardy et al. 2008). This activity is due to the presence of 11β-HSD1 in synovial fibroblasts. This activity is higher basally in synovial fibroblasts than in fibroblasts derived from other tissues such as lymph node or skin but activity in fibroblasts generated from all these tissues increases substantially in response to inflammatory cytokines (Hardy et al. 2006). The amount of glucocorticoid activating capacity observed in synovial tissue obtained from patients with rheumatoid arthritis correlated with systemic markers of inflammation. In both isolated fibroblasts and in synovial tissue 11β-HSD1 activity was able to suppress IL-6 production, an effect that could be blocked by a specific 11β-HSD1 inhibitor. Thus, 11β-HSD1 activity within synovium has the capacity to modify the local inflammatory response. These high levels of glucocorticoids are likely to have a Cushingoid like effect on nearby bone which is likely to exacerbate inflammation related bone loss. This may be a contributing factor in the development of the characteristic periarticular bone loss seen in inflammatory arthritis.
The role of 11β-HSDs in other tissue specific inflammatory diseases may be very different. In experimental colitis, 11β-HSD1 is also upregulated in colonic mucosa (Bryndova et al. 2004) and similar findings are observed in tissue obtained from patients with inflammatory bowel disease (Stegk et al. 2009). This effect is likely to increase the sensitivity of the colon to therapeutic glucocorticoids such as prednisolone. A high 11β-HSD1 activity in the inflamed colon may thus lead to more effective anti-inflammatory effects on the colon enabling a lower level of glucocorticoids to be used. This could have a potential bone sparing effect. The effects of 11β-HSD1 in modulating bone disease thus will depend on how this enzyme activity interacts with the response of the underlying disease to endogenous or therapeutic glucocorticoids. This factor is likely to impact on the ability of measures of 11β-HSD1 to predict the responses of bone to glucocorticoids in individuals with inflammatory disease.
Other therapies
Other therapies used in the treatment of inflammatory disease can themselves directly affect bone. Methotrexate and cyclosporine are both used as steroid-sparing agents in a range of conditions. Methotrexate appears to have no significant negative effect on bone but cyclosporine may have a mild negative effect (Abdelhadi et al. 2002, Minaur et al. 2002). These effects are difficult to study clinically since patients have normally previously been, or are concurrently treated with, glucocorticoids (Monegal et al. 2001). A further complicating factor is that these treatments alter the underlying inflammatory condition which is a critical factor in the bone loss itself. An example of such a treatment is anti-TNF therapy. This powerful anti-inflammatory medication has proven to be highly effective in many patients with inflammatory arthritis or inflammatory bowel disease. This treatment improves the underlying condition but also appears to have an independent beneficial effect on bone. This is primarily through inhibition of the ability of TNF-α to induce osteoclastogenesis. This may partly explain why anti-TNF treatment can protect against the development of bone erosions, an effect that may be independent of inflammation itself (Vis et al. 2006). An analysis of periodontal bone loss in patients with gingivitis receiving anti-TNF treatment is instructive (Pers et al. 2008). These patients experienced very little deterioration in periodontal bone damage during their anti-TNF treatment compared with similar patients not receiving anti-TNF. The treatment was, however, associated with a worsening of the underlying gingivitis presumably as a result of an impaired antibacterial immune response. The lack of deterioration in bone structure despite an increase in the underlying gingivitis reinforces the independence of bone resorption from inflammation itself. In animal models, similar phenomena are observed when inhibitors of the RANKL pathway are administered to mice undergoing experimental arthritis. These drugs are able to block bone and joint deterioration without any effect on the underlying inflammatory response (Kong et al. 1999a). Similar findings have been obtained in humans where a monoclonal antibody to RANKL has been shown to reduce the development of erosions in patients with rheumatoid arthritis, independent of any effect on inflammation (Cohen et al. 2008). These observations have led to a re-evaluation of treatment goals with awareness that suppression of inflammation and of bone and joint damage are interrelated but ultimately distinct objectives of treatment.
Sex steroid deficiency
It is well established that gonadal steroids have an important impact on bone (Eastell 2005). The bone loss seen after menopause or gonadectomy is primarily due to an increase in RANKL expression by osteoblasts and the corresponding increase in osteoclastogenesis (Hofbauer et al. 2000). Thus, normally oestrogen and testosterone have a tonic effect to restrain osteoclastogenesis. Inflammatory states are frequently complicated by centrally mediated hypogonadism that could have a negative impact on bone. The relative importance of this effect is difficult to distinguish from the other factors that underlie inflammation related bone loss. As described above, in the context of anorexia nervosa sex steroid replacement does little to improve bone health suggesting that other factors are involved such as the commonly observed reduction in circulating IGF-1 levels or a direct impact on bone of the central pathways that lead to hypogonadism (Miller et al. 2006). Although glucocorticoids can decrease sex steroid levels the adverse effects of glucocorticoids are still observed in patients with maintained menstrual cycle activity implying that the adverse effects of glucocorticoids are largely independent of hypogonadism (Chiodini et al. 1998). Studies in rodent models have reinforced this finding since glucocorticoid-effects on bone are additional to, but not mimicked by, ovariectomy (Weinstein et al. 2004).
Deficiency in other sex steroids has also been implicated in inflammation induced bone loss. The adrenal androgen dehydroepiandrosterone sulphate (DHEAS) is the most abundant steroid produced by the adrenal cortex and acts as a weak androgen. Inflammation leads to a dramatic reduction in DHEAS levels and this could lead to a further detrimental effect on bone health (Beishuizen et al. 2002). However, more recently, the role of DHEAS as a hormone has been questioned and it appears that the non-sulphated DHEA may be the active hormone in most situations (Arlt et al. 2006). The levels of DHEA do not decrease during inflammation but may actually increase with stress (Arlt et al. 2006). The role of DHEA replacement in inflammatory bone loss has not been examined but DHEA replacement in patients with idiopathic osteoporosis appears only to have a small effect in preserving bone density (Nair et al. 2006).
Immobility
Immobility has a dramatic effect on bone mediated by uncoupling of bone formation from resorption. This is seen in a variety of situations and can be a local (e.g. in paralysis) or systemic phenomenon. The effects of immobility have implications for all inflammatory diseases but especially neuromuscular and joint disease. The ways in which mechanical forces affect bone are still poorly understood. Bone loss is characterised by reduced formation and increased resorption (Carmeliet & Bouillon 1999). It is thought that osteocytes, cells that are found within bone matrix, mediate mechanosensing. This is likely to be through modulation of the other major pathways that couple bone formation and resorption such as the wnt pathway discussed above (Bonewald & Johnson 2008). This effect may partly be dependent on oestrogen receptor signalling suggesting that mechanosensing may be modulated by hypogonadism (Armstrong et al. 2007). Strategies to try to maintain forces on bone include use of exercise to control bone loss. A more sophisticated approach could see applied mechanical stimulation through administration of a vibration signal to induce an anabolic response to bone. This has not yet been explored in the setting of chronic illness.
Implications for therapy
Inflammation related bone loss is an important clinical problem and several approaches have been used in its prevention and treatment. One approach is to try to reverse the underlying mechanisms that cause the bone loss. Exercise and improved nutrition would seem to be logical treatments where reduced mobility and poor nutrition are implicated. Where exposure to therapeutic glucocorticoids is implicated, it would seem logical to try to reduce the dose given or use alternative anti-inflammatory agents. Unfortunately, these manoeuvres are usually not possible due to the nature of the underlying disease. The greatest success in treating inflammation related bone loss has come from manipulating the bone remodelling cycle independent of the underlying cause of the remodelling abnormality. In this approach, the relationship between bone formation and resorption and of the amount of bone turnover in general are manipulated such that any negative balance in favour of excessive resorption is abrogated and, ideally, the situation reversed is in favour of net bone formation. The most widely used treatments in this setting are bisphosphonates. These are osteoclast toxins that potently reduce bone resorption (Russell et al. 2008). This prevents excessive bone loss and also temporarily creates an imbalance where bone formation continues despite low resorption. Eventually, there is a secondary decrease in formation as would be expected from the coupling between resorption and formation. This approach works well in conditions where bone resorption is high but importantly also works in situations where the primary problem is a decrease in bone formation (Sambrook 2000). This is thought to be due to normalisation of the balance between formation and resorption in the setting of reduced overall bone turnover. A more recent approach is the use of agents that stimulate bone formation. The only widely used agent with such an effect is PTH and its analogues (Sambrook 2007). When given intermittently, subcutaneous PTH causes a dramatic increase in bone formation that exceeds the stimulation of bone resorption. This anabolic action of PTH is only seen when given in a pulsatile manner, continuous PTH exposure normally has a negative effect as described above (Poole & Reeve 2005). The theoretical benefit of these anabolic therapies is their capacity to reverse longstanding damage to the skeleton. However, clinical experience has shown that after an initial anabolic phase the balance between formation and resorption normalises with a corresponding reduction in the ability of these medications to be anabolic. This phenomenon is again likely to be due to the effects of the intrinsic mechanisms within bone to couple formation with resorption. Despite this limitation, there are other potential advantages of intermittent PTH during inflammatory disease. This treatment has been reported to protect osteoblasts against glucocorticoid-induced apoptosis (Jilka et al. 1999) and is clinically superior to antiresorbative therapy in patients with inflammatory disease treated with glucocorticoids (Saag et al. 2007). The beneficial effects have also been linked to activation of wnt signalling, inhibition of which is of potential importance in inflammatory bone loss in various ways (as described above).
At the moment therapies, the best are mildly anabolic but primarily work by being anticatabolic. The improved understanding of how bone remodelling is regulated raises the prospect that therapies that can uncouple bone formation from resorption in favour of formation for prolonged periods of time could be developed for use in the setting of inflammatory bone disease.
Conclusion
This review illustrates that multiple processes are involved in inflammation associated bone loss. These mechanisms appear to interact in complex ways and tend to reinforce each other through changes that impact on the bone remodelling cycle. An improved understanding of these mechanisms and how they might be modulated in a therapeutic setting is likely to lead to improved ways to reduce bone damage in chronic inflammatory disease.
Declaration of interest
There is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by a GlaxoSmithKline Clinician Scientist Fellowship to M C and an Arthritis Research Campaign Project Grant (number 18081).
- Received in final form 16 February 2009
- Accepted 24 February 2009
- © 2009 Society for Endocrinology










