
PHLPP: a putative cellular target during insulin resistance and type 2 diabetes
-
 Figure 1
Figure 1The three PHLPP isoforms share a common N terminal PH domain (pleckstrin homology), a series of leucine-rich repeats (LRR), a protein phosphatase domain and a PDZ binding motif (post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (DlgA) and zonula occludens-1 protein (zo-1) binding motif toward the C terminal. The splice variant PHLPP1β has an additional Ras association domain at its N-terminus. Its structure resembles with that of PHLPP2. A full colour version of this figure is available at http://dx.doi.org/10.1530/JOE-17-0081.
-
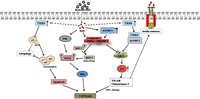 Figure 2
Figure 2Regulation of cell death by PHLPP involves parallel action of several factors. Absence of insulin or growth factors promotes phosphorylation of FOXO transcription factors by AKT, JNK and MST1 at conserved sites that allows causing cell death through bim induction. PHLPP dephosphorylates MST1 that activates FOXO and induces cell death progression during hyperglycemic stress. ROS-mediated TRB3 induction sequesters mTORC2 from phosphorylating AKT and thereby inhibiting its signaling. Simultaneously, hyperactivation of mTORC1 allows enhanced PHLPP1/2 expression leading to total AKT inhibition and insulin resistance. TRB3 further prevents p62/LC3 association, impeding autophagy. Further, p53 worsens insulin signaling by modulating the gluco-metabolic genes (TIGAR and glutaminase 2) along with the inhibition of mTORC2 and IGF-1/AKT axis through indirect intervention of PTEN, TSC1/2, SESTRIN and PTEN. Overall, the figure describes the putative mechanisms that promote apoptosis during insulin resistant states. A full colour version of this figure is available at http://dx.doi.org/10.1530/JOE-17-0081
-
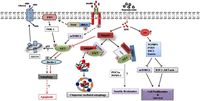 Figure 3
Figure 3The adaptor proteins FKBP51 and SRPK1 recruit PHLPP to elicit AKT dephosphorylation at S473 and allow the progression of insulin resistance through molecular intervention of FOXOs and PGC-1α with PHLPP. However, the exact mechanism remains incompletely understood. Aberrant autophagy, observed during hyperglycemic states, is regulated by PHLPP by checking beclin-1 phosphorylation via AKT inhibition and thereby instigating the cell to adopt apoptotic cell death. Further, the mTORC2/PHLPP/AKT complex controls chaperone-mediated autophagy regulating cell death. Excessive ROS induces p53 induction whose dynamic control over the glucose metabolic pathway and cell survival signaling apoptosis through inhibition of the IGF-AKT-mTORC2 axis. A full colour version of this figure is available at http://dx.doi.org/10.1530/JOE-17-0081.
-
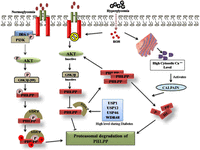 Figure 4
Figure 4During normoglycemic conditions, activated AKT allows bTrCP-mediated PHLPP ubiquitination. Phosphorylation of PHLPP1 by casein kinase 1 and GSK3β in the residues residing into the PP2C domain generates a phosphodegron motif that promotes PHLPP degradation. During hyperglycemia, activated GSK3β prevents phosphorylation of PHLPP1 thereby restricts its degradation leading to its constitutive stability. ER stress and mitochondrial dysfunction allow leaky release of calcium ions which promotes activation yet does not degrade PHLPP during hyperglycemia. Coexisting deubiquitinases further fine tune PHLPP degradation. Hence, several concomitant processes run to regulate PHLPP.
- © 2017 Society for Endocrinology









