Familial testicular germ cell tumors in adults: 2010 summary of genetic risk factors and clinical phenotype
- Mark H Greene,
- Christian P Kratz,
- Phuong L Mai,
- Christine Mueller,
- June A Peters,
- Gennady Bratslavsky1,
- Alex Ling2,
- Peter M Choyke3,
- Ahalya Premkumar2,
- Janet Bracci4,
- Rissah J Watkins4,
- Mary Lou McMaster5 and
- Larissa A Korde
- Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, Maryland 20852, USA
1Urologic Oncology Branch, National Cancer Institute, Center for Cancer Research, Bethesda, Maryland 20852, USA
2Diagnostic Radiology Department, National Institutes of Health, Warren G. Magnuson Clinical Center, Bethesda, Maryland 20892, USA
3Molecular Imaging Program, National Cancer Institute, Bethesda, Maryland 20852, USA
4Westat, Inc., 6116 Executive Boulevard, Suite 400, Rockville, Maryland, USA
5Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, Maryland 20852, USA
- (Correspondence should be addressed to M H Greene; Email: greenem{at}mail.nih.gov)
Abstract
Familial aggregations of testicular germ cell tumor (FTGCT) have been well described, suggesting the existence of a hereditary TGCT subset. Approximately 1.4% of newly diagnosed TGCT patients report a positive family history of TGCT. Sons and siblings of TGCT patients have four- to sixfold and eight- to tenfold increases in TGCT risk respectively. Segregation analyses suggest an autosomal recessive mode of inheritance. Linkage analyses have identified several genomic regions of modest interest, although no high-penetrance cancer susceptibility gene has been mapped yet. These data suggest that the combined effects of multiple common alleles, each conferring modest risk, might underlie familial testicular cancer. Families display a mild phenotype: the most common number of affected families is 2. Age at diagnosis is 2–3 years younger for familial versus sporadic cases. The ratio of familial seminoma to nonseminoma is 1.0. FTGCT is more likely to be bilateral than sporadic TGCT. This syndrome is cancer site specific. Testicular microlithiasis is a newly recognized FTGCT component. Candidate gene-association studies have implicated the Y chromosome gr/gr deletion and PDE11A gene mutations as genetic modifiers of FTGCT risk. Two genomewide association studies of predominantly sporadic but also familial cases of TGCT have implicated the KIT-ligand, SPRY4, and BAK1 genes as TGCT risk modifiers. All five loci are involved in normal testicular development and/or male infertility. These genetic data provide a novel insight into the genetic basis of FTGCT, and an invaluable guide to future TGCT research.
Background
There are three major subsets of testicular germ cell tumors (TGCTs): 1) the teratomas and yolk-sac tumors that occur primarily in neonates and infants; 2) pure seminoma, and a group termed collectively as nonseminoma – these tumors occur predominantly in young men; and 3) spermatocytic seminoma, which is typically diagnosed in older men (reviewed in Oosterhuis & Looijenga (2005)). Patients with familial TGCT (FTGCT) typically fall into category 2, which is by far the most common form of TGCT.
Among all malignancies, TGCTs are rare, accounting for ∼1% of all cancers in males (American Cancer Society 2009). They comprise the most common cancer in men aged 20–35 years. The rational, evidence-based development of effective therapy for both local and advanced TGCTs during the past 30 years represents one of the signal victories of modern oncology, with current cure rates exceeding 95%.
However, TGCT survivors are still at risk of the delayed toxicities from therapy, and men with a history of testicular cancer have a 40–65% increase in their risk of subsequent nongerm cell malignancy (Travis et al. 1997). In addition, the risk of developing cardiovascular disease is more than twice as high among testicular cancer treatment survivors relative to age-matched controls (Huddart et al. 2003). Other therapy-related complications include nephrotoxicity, neuropathy, and ototoxicity due to cisplatin, as well as pulmonary toxicity from bleomycin (Kollmannsberger et al. 1999). Post-treatment sexual dysfunction is not uncommon (van Basten et al. 1997, Huddart et al. 2005). Thus, although TGCTs are rare, affected young men are at risk of significant morbidity and mortality during their most productive years; disease-related medical and social costs are disproportionately greater than their frequency.
In the United States, 8400 new cases of TGCT were predicted to occur in 2009, with 380 deaths attributable to this disease (American Cancer Society 2009). In addition, 2–5% of patients develop cancer of the contralateral testicle (Wanderas et al. 1997), while 5–10% of patients fail the best available treatments, and ultimately die of their disease. Epidemiologic studies have documented an increasing incidence of TGCT since the mid-20th century (Huyghe et al. 2003, Purdue et al. 2005, Holmes et al. 2008). Between 1973 and 1995, the incidence of testicular cancer increased by 50% in the US. This increase in incidence correlates with the year in which patients were born, indicating a birth cohort effect (McKiernan et al. 1999). There are three incidence peaks in white males: the first occurring in childhood, with most cases representing rare, nongerm cell histologies; the second (and largest) appearing between ages 20 and 35 years (seminoma and nonseminoma histologies); and the third manifesting in the mid-60–70s, and comprising primarily of spermatocytic seminoma. The lifetime risk of TGCT in Caucasian men is estimated to be 1 in 230. It is the most common cancer in young white men aged 20–35 years, but it is significantly less common in African-Americans for reasons that are not well understood (SEER 1993, Swerdlow 1993).
Risk factors for TGCT are heterogeneous, with some – contralateral testicular cancer (Wanderas et al. 1997), cryptorchidism (Swerdlow et al. 1997b), persistent Müellerian duct syndrome (Duenas et al. 2001), testicular dysgenesis (Skakkebaek et al. 2003), infertility (Moller 1998, Jacobsen et al. 2000, Hotaling & Walsh 2009, Walsh et al. 2009), testicular atrophy (Dieckmann & Loy 1996, Moller et al. 1996), testicular intraepithelial neoplasia (Berthelsen et al. 1982, Skakkebaek et al. 1982, Dieckmann & Loy 1996), mixed gonadal dysgenesis (Kulkarni et al. 1990, Chemes et al. 2003), Klinefelter's syndrome (Smyth & Bremner 1998), and testicular microlithiasis (de Gouveia Brazao et al. 2004, DeCastro et al. 2008) – being more solidly established than others – inguinal hernia and hydrocele (Moller et al. 1996), polythelia (accessory nipples) (Goedert et al. 1984), in utero exposure to diethylstilbestrol (Goedert et al. 1984, Weir et al. 2000), gynecomastia (Olsson et al. 2002), and occupational exposure to polyvinyl chloride (Hardell et al. 1997).
With that by way of background, let us now turn our attention to familial and genetic aspects of testicular cancer in adults. The following represents a compilation of data obtained from 1) the peer-reviewed scientific literature, 2) the International Testicular Cancer Linkage Consortium (ITCLC), and 3) the NCI Clinical Genetics Branch (CGB) multidisciplinary etiologic study of FTGCT (NCI Protocol 02-C-0178; NCT 00039598).
FTGCTs: does a site-specific genetic syndrome exist?
Presently, when one thinks of familial cancer syndromes, those that come to mind are the disorders for which the genes have been discovered during the past several decades, and which, as a consequence, have been the focus of much discussion regarding their etiology, natural history, and management (Lindor et al. 2008), such as the hereditary syndromes involving breast/ovarian cancer and colorectal cancer, as well as multiple endocrine neoplasia type 1 (MEN1) and type 2 (MEN2). These disorders are all caused by rare germline mutations in highly penetrant genes, and are associated with dramatically affected pedigrees involving many cases over multiple generations, with earlier-than-usual onset of disease and a tendency to bilateral malignancy when paired organs are at risk. It is less widely appreciated that nearly all cancers include a small subset of patients who display familial aggregation of disease (Lindor et al. 2008), although the genetic basis for these clusters remains unknown for many of them, as is the case for FTGCT.
The first level of evidence suggesting the presence of a hereditary form of TGCT comprises numerous scientific reports of multiple-case families (reviewed in Kramer & Greene (2005)). This historical information has recently been supplemented by a descriptive analysis of 461 kindred assembled by the ITCLC as part of its gene discovery activities (Mai et al. 2009b). A meta-analysis indicates that an estimated 1.4% of men with newly diagnosed TGCT report a positive family history for this cancer (Dieckmann & Pichlmeier 1997). For the moment, suffice it to say that the case-report literature provides ample evidence to suggest that a hereditary form of TGCT might exist.
The next level of evidence derives from the epidemiologic literature, in which a family history is often ascertained (see Introduction to Lindor et al. (2008)). In general, the familial relative risks for most of the common adult cancers are in the range of 1.5–2.5 for the first-degree relatives of the cases. Similar analyses of testicular cancer have shown that sons of men with TGCT have a four- to sixfold increase in TGCT risk, while siblings of men with TGCT have an eight- to tenfold increased risk (Dong & Hemminki 2001, Hemminki & Li 2004). These are among the highest familial relative risks reported for any human cancer, and they provide the first real quantitative evidence in support of a genetic hypothesis for FTGCT. Moreover, a 37-fold/76.5-fold elevated risk of TGCT in dizygotic/monozygotic twin brothers of men with TGCT has been reported (Swerdlow et al. 1997a).
Finally, segregation analysis represents the most quantitative method discussed thus far. This is an analytic tool for modeling the distribution of affected and unaffected individuals in a family or set of families, and testing whether the observed distribution of cases is statistically consistent with a definable Mendelian mode of inheritance, a polygenic model or an environmental model. These studies are methodologically difficult to perform properly, and there is only one study which has been performed to date on series of FTGCT kindred (Heimdal et al. 1997). In this analysis, an autosomal recessive model provided the best fit with the data, but it was not particularly robust primarily due to the relatively small numbers of families available for analysis. However, this observation nonetheless provides another quantitative set of data in support of the genetic hypothesis. Notably, an autosomal recessive model was also suggested by Nicholson and Harland, who performed a statistical analysis of published data on the age of onset of TGCT and of the prevalence of bilateral disease in familial and general cases (Nicholson & Harland 1995).
Based on all the above data in the aggregate, it is reasonable to conclude that there is a familial variant of TGCT, but whether it truly has a genetic basis or not remains to be determined. In familial disease research, one must keep in mind that genes are not the only factors that cluster in families (e.g. intra-familial diets tend to be similar, there may be shared environmental/occupational exposures, similar patterns of reproductive decision making, etc.), so all that is familial is not necessarily hereditary. We will return to the question of testicular cancer susceptibility genes shortly.
What resources are required for new gene discovery in FTGCT?
Historically, collecting a large series of multiple-case families upon which to focus a detailed, in-person, etiologically oriented clinical research assessment has proven to be an efficient strategy for studying poorly defined familial disorders. The CGB of the US National Cancer Institute implemented such a study targeting FTGCT in 2002 (NCI Protocol 02-C-0178; NCT 00039598). During the ensuing 7 years, we have accrued 122 multiple-case families with 608 consented study subjects, including 180 affected men. We have obtained blood samples from 547 subjects, and have examined 147 participants at the NIH Clinical Center. The goals of this project included a) accruing new families for gene discovery; b) defining the FTGCT clinical phenotype; c) performing psychosocial and behavioral research; and d) creating a carefully annotated biospecimen repository to support translational and etiologic research.
The DNA samples from these families were contributed to the gene discovery activity generated by the ITCLC (Leahy et al. 1995), an organization created to assemble a sufficiently large number of genetically informative, multiple-case TGCT families to permit mapping and cloning potential testicular cancer susceptibility genes. The CGB FTGCT study complemented the ITCLC research agenda, since collection of DNA samples rather than individual etiologic risk assessment was the primary focus of the former.
Thus, the CGB project has been able to perform a variety of studies that were not feasible in the context of a large, multi-center international study. These have included seeking the presence of a dysmorphic clinical phenotype (null), identifying unique clinical constellations suggestive of novel syndromes (Mai et al. 2007), quantifying the prevalence of genitourinary anomalies, searching for constitutional cytogenetic abnormalities (see below), evaluating interest in genetic testing (Peters et al. 2006) and general genetic knowledge (Peters et al. 2008), studying cancer screening behavior related to early detection of testicular (Vadaparampil et al. 2009) and other cancers, evaluating patterns of reproductive decision making and issues related to fertility, collecting semen samples for translational research projects, and demonstrating that net endogenous DNA damage levels are higher in FTGCT patients who develop nonseminoma compared with seminoma (Cook et al. 2008). The latter finding may partly explain the more aggressive biology and younger age-at-onset of nonseminoma GCTs compared with the relatively less aggressive, later-onset seminomas. We also reported that ovarian germ cell tumors might be part of the cancer phenotype within multiple-case testicular cancer families (Giambartolomei et al. 2009).
Thus, a comprehensive approach aimed at understanding poorly defined familial cancer syndromes can benefit from both single-institution and multi-institution, international research strategies.
What is the FTGCT phenotype?
In order to attempt finding a gene by conventional linkage analysis, a precise definition of the disease phenotype is essential. Based on the literature, and the recently published ICTLC descriptive analysis, here is what we currently know. The ITCLC data set consisted of 985 FTGCT cases from 461 families contributed by 18 international centers (Mai et al. 2009b). The vast majority (88%) of families consisted of two and only two cases; the maximum number of affected members observed in a single family was 5, and only two such kindred were detected. Thus, these families differed dramatically from the more recently studied susceptibility disorders such as BRCA-related hereditary breast/ovarian cancer, in which multiple-case, multi-generation families were commonly observed. This suggests that the hereditary FTGCT trait, if there is one, is associated with a very low penetrance. This affection pattern also creates serious constraints on the statistical power of linkage analysis as a gene-finding strategy.
Further complicating matters, multiple potential patterns of inheritance were seen from family to family, including those consistent with autosomal recessive (49% are sibling pairs), autosomal dominant (father/son, grandfather/father/son: 19%) and X-linked inheritance (absence of male-to-male transmission), as well as more complex patterns involving aunts, uncles, and cousins. This raises the very real possibility of genetic heterogeneity within the overall entity which, if true, further diminishes the statistical power of linkage analysis.
In a subset of ITCLC families (drawn from centers with large numbers of families and the availability of a national cancer registry), age at diagnosis was statistically significantly younger for FTGCT cases from North America (P=0.024), the United Kingdom (P<0.0001), and Australia/New Zealand (P=0.0033) compared with general population cases. When stratified by histology, the difference in age-at-diagnosis distribution between familial and population cases was observed for seminoma cases from North America (P=0.002) and the United Kingdom (P<0.0001), and for nonseminoma cases from the United Kingdom (P=0.029) and Australia/New Zealand (P=0.0023). Overall, the mean age at FTGCT diagnosis was about 2.5 years younger than that observed for sporadic TGCT from the same country. Although these differences were statistically significant, and they fit with the general notion that hereditary cancers typically display a younger-than-usual age at cancer diagnosis, they were not large enough to identify a subset of multiple-case families as either unusually early or late onset (Mai et al. 2009a).
Approximately 6.5% of FTGCT cases reported in the ITCLC series had both testes affected (i.e. bilateral cancer), a prevalence somewhat greater than that observed in sporadic TGCT. The prevalence of cryptorchidism (21% of families; 14% of familial cases) was similar to that seen in sporadic TGCT. Both testes were equally likely to be affected, and there were no correlations in tumor histology, either within families or among men with bilateral disease (Mai et al. 2009b).
One of the main goals of this analysis was to identify one or more characteristics that might define a more homogeneous set of families, making it easier to detect a linkage signal. Families with affected cousins differed from the other families on several of these variables but, as a consequence of multiple testing, this could be a false positive finding (Mai et al. 2009b). In an analogous strategy, efforts are currently underway to quantitatively evaluate the risk of cancers other than germ cell tumors to see if the spectrum of syndrome-related malignancies might be broader than expected. We are also conducting a blinded central pathology review of both familial and sporadic TGCTs by expert germ cell tumor pathologists in the hope of identifying subtle differences in these histologically complex tumors that might provide a guide to sub-group homogeneity in a manner similar to that applied so successfully in the hereditary renal cancer syndromes (Linehan et al. 2009).
The recognition that testicular microlithiasis has a particularly strong association with FTGCT represents a novel finding that might reflect underlying syndromic heterogeneity. Microlithiasis is gaining acceptance as a risk factor for sporadic testicular cancer (Parenti et al. 2007), although its precise role in this context has yet to be fully defined (DeCastro et al. 2008). Its potential contribution to FTGCT has been reported recently by both UK (Coffey et al. 2007) and US (Korde et al. 2008) investigators. The former recruited both familial and sporadic TGCT patients as well as unaffected male relatives and healthy male controls (total=328 subjects). Testicular microlithiasis was observed more frequently in TGCT cases than in controls (36.7 vs 17.8%; P<0.0001) and in unaffected male relatives than in controls (34.5 vs 17.8%; P=0.02). TGCT cases and matched relative pairs were significantly concordant for microlithiasis (P=0.05).
Our study targeted 81 men (48 with TGCT and 33 without TGCT) from 31 extended multiple-case families (Korde et al. 2008). Testicular microlithiasis was more frequently observed in the contralateral testicles of affected men than in those without cancer (48 vs 24%; P=0.04). Testicular microlithiases were bilateral in 87% of the unaffected men. Ten of the 31 families accounted for a majority (61%) of microlithiasis cases. The presence of microlithiasis was not correlated with tumor histology, testicular maldescent, inguinal hernia, history of testicular injury or infection, or treatment of TGCT. This report also includes the first documented incident TGCT diagnosed during prospective follow-up of a family member with microlithiasis. Both studies employed the same microlithiasis definition, i.e. either ‘classic’ (≥5 microliths), or ‘limited’ (<5 microliths).
In the aggregate, these two reports document that testicular microlithiasis is significantly more common among FTGCT family members than in the general population (range 0.6–9.0%), and is more common among FTGCT cases versus their unaffected blood relatives. It also appeared to cluster in certain families. These findings suggest both a familial predisposition to testicular microlithiasis and an association between microlithiasis and FTGCT. If further corroborated, the presence of microlithiasis could signal a need for more intensive surveillance and, perhaps, early testicular biopsy in a subset of FTGCT family members. Stratifying genetic linkage analyses by the presence or absence of microlithiasis in family members might improve statistical power by focusing on etiologically distinct, genetically more homogeneous family sets (see below for more information on this issue).
What has been learned from linkage analysis in the ITCLC family set?
The ITCLC was formed to permit enrolling sufficient numbers of multiple-case TGCT kindred required to achieve the statistical power needed to map and clone one or more susceptibility genes. The first linkage analysis, published in 2000, appeared to validate this approach (Rapley et al. 2000), when a strong linkage signal was detected at chromosome Xq27. This study included 134 families with ≥2 TGCTs, 99 of which were consistent with X-linkage. The odds in favor of linkage (i.e. the HLOD score) in this subset was 2.01, and if X-linked families which contained at least one family member with bilateral TGCT were targeted, the HLOD score rose to 4.70 (generally, HLOD scores >3.0 are considered a strong, statistically significant linkage signal). This finding led to a major effort aimed at recruiting additional families in order to validate this observation.
Three years later, a second report expanded this series to 178 families (including the original 134), with an analytic focus on autosomal genes (Rapley et al. 2003). That analysis yielded an HLOD score of 2.05 for chromosome 12q12–q13, the same region that commonly displays somatic cytogenetic rearrangements in testicular tumor tissue.
The most recent Consortium analysis was published in 2006, and it included a further expansion of the study set to 237 families, of which 163 families were compatible with X-linkage. In that analysis, the aggregate Xq27 HLOD score dropped to 1.01 (Crockford et al. 2006), substantially weakening, but not definitively eliminating, the putative association between Xq27 and FTGCT. This analysis also detected modest linkage signals (HLOD>1.0) at several autosomal sites, including 2p23; 3p12; 3q26 (LOD=1.94); 12p13–q21; and 18q21–q23. Of note, the LOD score for the chromosome 12 locus detected in 2003 declined in this larger data set as well. These data were interpreted as inconsistent with a single, rare, highly penetrant susceptibility gene model. Rather, it seemed likely that the combined effects of multiple common alleles, each conferring modest risk, might explain the familial aggregation of testicular cancer.
What does one do when linkage analysis fails?
Fortunately, there are alternative gene discovery strategies that can be deployed.
Cytogenetic analysis
One option which has proved to be successful in other familial cancer syndromes (e.g. retinoblastoma, Wilms tumor, and familial adenomatous polyposis) is to search for constitutional cytogenetic abnormalities in the hope that finding chromosomal breakpoints might prove to be genomic regions of etiologic interest. Using high-resolution Giemsa-banding and spectral karyotyping, we studied 28 affected individuals from 17 independent multiple-case TGCT families. We found no disease-associated germline cytogenetic abnormalities either in our case series or in 17 previously reported cases (Mueller et al. 2007). Studies of this kind, while worth attempting, require a great deal of luck to succeed. We have opted not to pursue this avenue of investigation further.
Candidate gene analysis
Until recently, ‘candidate gene analysis’ has been the most common alternate discovery strategy, in which genes that are thought to be biologically plausible candidates are systematically analyzed in search of germline mutations that might explain disease risk. This is an arduous task, given that the human genome contains more than 25 000 genes, and that recent experience has shown that we are not always correct when it comes to selecting biologically plausible candidate genes for a particular disease. And, we are learning with increasing frequency that some very important susceptibility genes have biological functions that would not be intuitively associated with particular diseases.
That said, the candidate gene strategy has been deployed in FTGCT, with mixed results. First, three genes (FMR1, CXorf1, and the hypothetical gene LOC158813/158812) known to reside in the Xq27 locus identified by the first linkage analysis have been studied in this fashion by investigators at the Institute of Cancer Research in the United Kingdom, with no success (Crockford et al. 2006). Secondly, based on the frequent detection of somatic mutations in the KIT oncogene in human testicular cancers, the ITCLC screened DNA from 240 multiple-case testicular cancer families, and no germline mutations were detected (Rapley et al. 2004).
Thirdly, germline mutations in the mouse Dnd1 gene have been shown to cause testicular tumors in mouse strain 129. Accordingly, ITCLC investigators analyzed the human ortholog of this gene, known as DND1, in 434 familial and 671 sporadic human TGCT subjects (Linger et al. 2008). Only a single rare variant was found (versus zero of 4132 control chromosomes). The authors concluded that this gene could, at the very best, make only a tiny contribution to the genetic etiology of FTGCT. This gene has not been studied further.
Fortunately, the candidate gene strategy has not been completely fruitless. ITCLC investigators decided to analyze the potential role of the Y chromosome gr/gr deletion as an FTGCT risk factor, recognizing that male infertility is an established risk factor for sporadic testicular cancer, and that the gr/gr deletion is the most commonly identified genetic cause of male infertility (Nathanson et al. 2005). The deletion was detected in 13 of 431 (3.0%) FTGCT cases, 28 of 1376 (2%) sporadic TGCT cases, and 33 of 2599 (1.3%) of unaffected males, corresponding to threefold (95% CI 1.5–6.7) and twofold (95% CI 1.3–3.6) increases in TGCT risk respectively. These findings, which comprise the first documented link between a specific genetic locus and the risk of FTGCT, indicate that the Y chromosome gr/gr microdeletion is a rare, low-penetrance TGCT susceptibility allele for both the familial and sporadic forms of TGCT. The biological plausibility of this result provides further support for the validity of these findings.
The most recent candidate gene study related to FTGCT targeted a phosphodiesterase gene known as PDE11A (Horvath et al. 2009), a critical regulator of cyclic AMP signaling in adrenal and other steroidogenic tissues, which is highly expressed in the normal human testis. Pde11a null mice are infertile (Wayman et al. 2005), and germline variations in this gene have been implicated in adrenal gland neoplasia (Horvath et al. 2006). We sequenced the PDE11A gene in 95 TGCT patients from 64 unrelated multiple-case kindreds, and identified eight nonsynonymous substitutions (four of which were unique to FTGCT subjects) in 20 patients from 15 distinct families. The prevalence of all PDE11A gene variants (combined) was significantly higher among patients with TGCT (P=0.0002) than among controls; they were detected in 19% of the families studied. Functional studies showed that all these mutations reduced PDE activity, and that PDE11A protein expression was decreased (or absent) in testicular tumor samples from mutation carriers. We concluded that PDE11A-inactivating sequence variants appear to modify the risk of FTGCT.
Genomewide association studies
Genomewide association studies (GWAS) comprise the newest arrow in the genomics analysis quiver (reviewed in McCarthy et al. (2008)). In contrast to the candidate gene strategy, GWAS is undertaken with no specific genetic target in mind (this approach is often designated ‘agnostic’), and it leverages the power of high-throughput genomic technologies to analyze 500 000 to 1 000 000 (or more) genetic variants in the form of single nucleotide polymorphisms (aka ‘SNPs’) on each of the thousands to tens of thousands of DNA samples. Associations between each variant and the disease of interest are sought, with the expectation that most statistically significant positive findings will represent a positive signal resulting from a disease-modifying gene at or near the implicated SNP. The details, strengths, and limitations of this analytic approach are beyond the scope of the current presentation. Suffice it to say that this technique has proven itself capable of identifying both novel genetic loci and genes thought to be biologically plausible as genetically important in a broad diversity of both neoplastic and nonneoplastic disorders. One of the major limitations that has emerged from this new body of work is the fact that most associations that have been identified to date are small in magnitude, i.e. odds ratios (ORs) of 1.2–1.6 predominate. Consequently, with a few rare exceptions, most of these findings have been difficult to translate directly into clinical practice, although they surely have opened many new doors into understanding disease mechanisms and pathogenesis.
This technology was recently applied to testicular cancer (largely sporadic) by British (Rapley et al. 2009) and American (Kanetsky et al. 2009) investigators. In the former two-stage study of 1301 TGCT cases and 3241 controls, a strong evidence for susceptibility loci was detected on chromosome 12 (per allele OR=2.55 (95% CI=2.05–3.19), P=10−31), chromosome 5 (per allele OR=1.37 (95% CI=1.19–1.58), P=3×10−13), and chromosome 6 (per allele OR=1.50 (95% CI=1.28–1.75), P=10−13). Individuals who were homozygous for chromosome 12 variant alleles had TGCT ORs of 6, currently the strongest association between any GWAS candidate and the risk of any cancer. KIT ligand (KITLG), which encodes the ligand for the receptor tyrosine kinase KIT, is the gene which likely explains the chromosome 12 association. This gene has previously been implicated in both the pathogenesis of sporadic TGCT and the normal biology of germ cells. Kitl homozygous null germline mutations in mice cause infertility due to progenitor germ cell development failure.
The SNPs implicated on chromosome 5 are physically very close to the SPRY4 gene, an inhibitor of the mitogen-activated protein kinase pathway, which is activated by the KITLG–KIT pathway, while those implicated on chromosome 6 are in a BAK1 (BCL2-antagonist/killer 1) gene intron. The KITLG–KIT pathway represses BAK1 expression in testicular germ cells, and is implicated in the germ cell apoptosis that occurs in response to blockade of this pathway. More detailed analyses revealed no differences in these associations when the data were stratified by tumor histology, cancer laterality (unilateral versus bilateral), or family history (n=220). The lack of an even stronger association among family history-positive cases was a surprise, and may have been due to the relatively small sample size of this subset, which comprised only ∼17% of the case set. Overall, these findings are remarkable for the strength/magnitude of the associations, for the biological coherence of the three implicated genes, and for their apparent link to TGCT risk through pathways involved in male infertility and normal germ cell development.
The second GWAS – comprised almost exclusively of family history-negative subjects – confirmed both the chromosome 12 (KITLG) and chromosome 5 (SPRY4) findings, despite its substantially smaller sample size (648 cases; 1779 controls) (Kanetsky et al. 2009). Independent corroboration of GWAS results is an essential component of this analytic strategy, since it provides considerable reassurance that findings are not a consequence of false discovery, which is one of the major theoretical pitfalls inherent in the huge number of statistical comparisons that are inevitably made in the course of such a study. The American investigators also offered a novel explanation of the known striking differences in TGCT incidence between whites (high) and blacks (very low) based on recognizing that KITLG plays an important role in skin pigmentation. Using information from the HapMap3 data set, they illustrated that the prevalence of the KITLG disease-associated variants was significantly higher in normal white versus normal black populations. It is plausible to hypothesize that the racial differences in TGCT incidence may, at least in part, be due to population differences in KITLG variant frequency.
The biology of the KITLG–KIT pathway and its relevance to testicular development
KITLG belongs to the family of short-chain helical cytokines. Two isoforms have been identified, consisting of soluble and transmembrane versions of this protein. KITLG initiates multiple cellular responses by activating its receptor KIT. The biologic function of the KIT signaling pathway is broad and includes development of hematopoietic cells, melanocytes, and germ cells (reviewed in Ashman (1999)). Notably, recent studies in mice demonstrate that Kitl controls midline cell death of primordial germ cells and is essential for their normal proliferation and migration (Runyan et al. 2006). Moreover, Kitl governs primordial germ cell survival and motility, and provides a continuous niche throughout their migration (Gu et al. 2009).
Mutations of KIT and KITLG have other associations with human disease. Germline mutations of KITLG cause familial progressive hyperpigmentation (Wang et al. 2009), while KIT germline mutations lead to familial gastrointestinal stromal tumors (Nishida et al. 1998) and familial diffuse cutaneous mastocytosis (Tang et al. 2004). Notably, somatic mutations (or overexpression) of the KIT oncogene are detected at various frequencies in several neoplasms, including both familial and nonfamilial testicular cancers (Rapley et al. 2004, Coffey et al. 2008). In this regard, it is notable that loss of the transmembrane Kitl isoform increases susceptibility to TGCT in a murine model (Heaney et al. 2008). Moreover, administration of the tyrosine kinase inhibitor imatinib (which includes KIT among its ligands) has resulted in complete response in pretreated disseminated testicular seminoma with KIT overexpression (Pedersini et al. 2007).
Elucidating the mechanisms through which the recently identified genetic SNPs affecting the KITLG–KIT pathway contribute to TGCT development has now emerged as a major scientific priority. It is conceivable that these variants deregulate the signaling pathway (e.g. by altered gene expression), and/or that they render testicular germ cells more susceptible to somatic mutations required for neoplastic transformation.
Summary of genetic studies to date
Segregation analyses support the existence of Mendelian inheritance in multiple-case TGCT families, with the evidence most strongly favoring an autosomal recessive mechanism. For the various reasons described above, traditional linkage analysis has not been as productive as we might have hoped relative to defining genomic regions of etiologic interest. Candidate gene analyses have had mixed results. Earlier studies found no convincing evidence to support an etiologic role for germline mutations in three Xq27 genes, KIT or DND1. Subsequent studies have implicated the Y chromosome gr/gr deletion and germline variants in the PDE11A gene as potential modifiers of FTGCT risk. And the most recent analyses, based on the GWAS strategy, have implicated KITLG, SPRY4, and BAK1 as sporadic testicular cancer susceptibility loci (see Table 1 for summary). It is noteworthy that the latter five loci have all been implicated in normal embryologic gonadal development and in male infertility. In the aggregate, these findings provide an evidence-based foundation for future genomic research. The strength of these associations (derived from studies of both familial and sporadic TGCTs), their biological coherence and plausibility, and the replicability of the GWAS results suggest that we are on the brink of new insights into the pathogenesis of TGCT. Figure 1 summarizes the factors that are known to be relevant in the pathogenesis of TGCT and FTGCT.
Genes and genomic regions implicated in familial testicular cancer
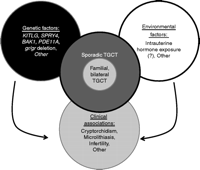
Factors contributing to the pathogenesis of testicular germ cell tumors. Several genes and genomic regions have been implicated in the pathogenesis of testicular germ cell tumors (TGCTs). Interestingly, all these loci have also been implicated in testicular development. While these variations explain a proportion of the genetic risk, the remaining genetic factors remain undetermined. Epidemiologic data strongly suggest that TGCTs have a significant environmental component. However, these external factors remain poorly defined. Some of the same factors that lead to TGCTs also underlie infertility, providing an explanation for the clinical association between TGCTs and infertility. Possibly, similar relationships are also true for microlithiasis and cryptorchidism. It is conceivable that the familial form of TGCTs is caused by the same factors that lead to sporadic TGCTs; however, we hypothesize that patients with the familial and/or bilateral form of TGCTs have a higher risk score (e.g. a larger number of risk factors).
Conclusion
A great deal of progress has been made toward elucidating both the clinical phenotype and the genetic basis of familial testicular cancer since the initial chromosome Xq27 linkage finding was published 10 years ago. These successes can, in large part, be attributed to the formation of the ITCLC, and the willingness of its members to accrue, enroll, and share their data derived from multiple-case TGCT families (Leahy et al. 1995, Rapley et al. 2000, 2003, 2004, 2009, Nathanson et al. 2005, Crockford et al. 2006, Linger et al. 2008, Mai et al. 2009a,b). This disorder is so rare that even the largest of the individual ITCLC centers would have found it impossible to conduct on its own the full range of clinical and laboratory research studies required to define the FTGCT phenotype and to characterize its genetic basis. The collection of families assembled by the ITCLC comprises a unique and invaluable resource. It provides a setting in which genetic studies performed in sporadic TGCT can be quickly and efficiently evaluated in high-risk families.
In this modern era of ‘big genomic science’, in which the primary research focus has shifted to large, international multi-institution analyses of pooled data sets comprising tens of thousands of study participants and literally millions of data elements, it is easy to lose sight of the fact that a great deal of important work can still be accomplished on a smaller scale, even by a single institution, in which study subjects are seen face to face, and evaluated in a comprehensive, systematic, multidisciplinary fashion. The NCI CGB Familial Testicular Cancer project is an excellent illustration of this research strategy (Peters et al. 2006, 2008, Mai et al. 2007, Mueller et al. 2007, Cook et al. 2008, Korde et al. 2008, Giambartolomei et al. 2009, Horvath et al. 2009, Vadaparampil et al. 2009), having yielded both novel, high-impact positive findings and definitive negative results, both of which are helping to shape ongoing and future research efforts into the etiology, clinical phenotype, and management of familial testicular cancer.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the Intramural Research Program of the US National Cancer Institute, National Institutes of Health, and by support services contracts NO2-CP-11019-50 and N02-CP-65504 with Westat, Inc., Rockville, MD, USA.
- © 2010 Society for Endocrinology










