Progesterone receptors act as sensors for mitogenic protein kinases in breast cancer models
- Department of Medicine (Division of Hematology, Oncology, and Transplantation) and Pharmacology, Masonic Cancer Center, University of Minnesota, MMC 806, 420 Delaware Street, Minneapolis, Minnesota 55455, USA
- (Correspondence should be addressed to C A Lange; Email: lange047{at}umn.edu)
Abstract
Progesterone receptors (PR), members of the nuclear receptor superfamily, function as ligand-activated transcription factors and initiators of c-Src kinase and mitogen-activated protein kinase signaling. Bidirectional cross-talk between PR and mitogenic protein kinases results in changes in PR post-translational modification, leading to alterations in PR transcriptional activity and promoter selectivity. PR-induced rapid activation of cytoplasmic protein kinases insures precise regulatory input to downstream cellular processes that are dependent upon nuclear PR, such as cell-cycle progression, and pro-survival signaling. Here, we review interactions between PR and mitogenic protein kinases and discuss the consequences of specific post-translational modifications on PR action in breast cancer cell-line models.
Introduction
Members of the steroid hormone receptor family act as ligand-activated transcription factors and function as direct activators of cytoplasmic signal transduction molecules. Genes transcribed by steroid receptors (SRs) encode a diverse array of proteins carrying out multiple cellular processes, including regulation of metabolism, cell-cycle progression, and survival. SR transcriptional activity has been well studied on artificial promoter–reporter genes, yet mechanisms of endogenous gene regulation and selectivity remain poorly understood. Recently, it has become well accepted that SRs also act in critical cytoplasmic intracellular signaling complexes, yet these so-called ‘rapid’ actions of SRs remain vastly understudied relative to their nuclear activities. The progesterone receptor (PR) is transcribed from a single gene via alternate usage of up to three independent translational start sites resulting in PR-A, PR-B, and PR-C isoforms (Kastner et al. 1990). PR-C appears to be a uterine-specific protein (Condon et al. 2006), while PR-A and PR-B are coexpressed in breast tissues. PR-B, the full-length form of the protein, has a molecular mass (MW) of 116 kDa, while PR-A (MW=94 kDa) lacks the N-terminal 164 amino acids. The region of PR-B upstream of the PR-A start site has been named the B-upstream segment (BUS) as this region is unique to PR-B (Fig. 1). PR-A and PR-B contain the critical components for nuclear receptor function: the ligand-binding domain; hinge region (H); DNA-binding domain (DBD); and two out of the three activating function domains. Although both PR-A and PR-B can activate gene transcription, they function as distinct and independent transcription factors, each capable of mediating specific transcriptional responses; PR-B is both a nuclear and cytoplasmic protein, and is thus available to activate cytoplasmic or membrane-associated signaling cascades (Boonyaratanakornkit et al. 2001, 2007).
Schematic of progesterone receptor and its phosphorylation sites. All three PR isoforms (PR-A, PR-B, and PR-C) are transcribed from the same gene, containing distal and proximal promoters, and created via differential use of two internal translational start sites. Shown are three transcription activation function (AF) domains, the B-upstream segment (BUS), the DNA-binding domain (DBD), the hinge region (H), and the hormone-binding domain (HBD). The progesterone receptor is phosphorylated basally, as well as in response to hormone. Shown here are the various sites of phosphorylation as determined in vitro and in vivo, and kinases that are likely responsible for phosphorylation at these sites.
PRs are considerably post-translationally modified by phosphorylation, sumoylation, ubiquitination (Weigel 1996, Lange et al. 2000, Abdel-Hafiz et al. 2002), and acetylation (unpublished data). Up to 14 residues in PR-B are known to be phosphorylated in vitro or in vivo (Fig. 1; Lange 2004). Phosphorylation of PR at specific sites can occur basally, upon ligand binding, and/or upon protein kinase activation in response to peptide growth factors. The phosphorylation state of PR may influence its subcellular localization (Qiu et al. 2003), transcriptional activity (Takimoto et al. 1996, Shen et al. 2001, Pierson-Mullany & Lange 2004, Narayanan et al. 2005b), rate of turnover (Takimoto et al. 1996, Weigel 1996, Lange et al. 2000), protein complex formation, and target-gene specificity (Takimoto et al. 1996, Weigel 1996, Lange et al. 2000, Qiu et al. 2003, Narayanan et al. 2005a, Boonyaratanakornkit et al. 2007, Daniel et al. 2007a, Faivre et al. 2008). Kinases known to phosphorylate PR include mitogen-activated protein kinase (MAPK), casein kinase II, and cyclin-dependent protein kinase-two (CDK2; Weigel et al. 1995, Weigel 1996, Shen et al. 2001).
PR interacts with c-Src and the MAP kinase module
PR contains several domains critical for rapid activation/interaction with cytoplasmic protein kinases (Fig. 2; Migliaccio et al. 1998, Boonyaratanakornkit et al. 2001, Ballare et al. 2003). In particular, liganded PR binds the SH3 domain of c-Src directly through a proline-rich domain located in its N-terminus (Boonyaratanakornkit et al. 2001). PR thus activates c-Src by engagement of its SH3 domain, resulting in the recruitment and activation of downstream ERK1/2 MAPK modules. The consequences of this rapid signaling event vary with cell type; T47D breast cancer cells exhibited increased proliferation, whereas MCF12A (untransformed, non-tumorigenic) breast cell proliferation was inhibited. Additional studies demonstrated that PR and estrogen receptor-α (ER) independently interact with the c-Src SH3 (PR) and SH2 (ER) domains.
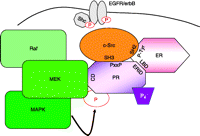
PR-scaffolding interactions. Previously reported interactions between PR (PxxP; proline-rich domain) and/or ER (phospho-tyrosine 537) and c-Src (SH3 domain with PR, SH2 domain with ER), as well as interactions between PR (ERID; estrogen receptor interaction domain) and ER (LBD, ligand-binding domain), have been shown to be necessary for progesterone-induced c-Src/MAPK activation (Arnold et al. 1995a,b, Migliaccio et al. 1998, Boonyaratanakornkit et al. 2001, Ballare et al. 2003). Additionally, complex formation between PR (CD domain) and MEK1 (D domain) may be necessary for MEK1 docking and subsequent PR post-translational modification and activation. These PR-scaffolding complexes are likely localized to the membrane through their interactions with Shc and EGFR (Song et al. 2007).
A second model of PR-induced activation of c-Src/MAPK requires the presence of ER (Migliaccio et al. 1998). Following treatment of T47D cells with synthetic progestin (R5020), activated c-Src and ERK2 were detected within 2–5 min. In contrast to studies described above, progestin-induced MAPK activation was blocked following treatment with anti-estrogens. Thus, ER is implicated in progesterone-dependent activation of MAPK. Further protein–protein interaction experiments using endogenous (in T47D cells) or exogenous (transfected into COS-7 cells) proteins demonstrated an interaction between c-Src, PR, and ER. In this model, ER appeared to be required for interaction of PR with c-Src, as well as subsequent activation of MAPK. The magnitude of progestin-induced MAPK activation in T47D cells was nearly identical to epidermal growth factor (EGF) stimulation and this was associated with an increase in cell proliferation. In a subsequent report, two regions within the PR N-terminus were found to directly interact with ER, ERID-I, and -II (ER-interacting domains I and II; Ballare et al. 2003). Ligand binding to PR induced an interaction between ER and the SH2 domain of c-Src leading to activation of MAPK (Ballare et al. 2003). Although this research group was unable to detect an interaction between PR and c-Src in vivo (Migliaccio et al. 1998), they detected direct interactions between these two proteins in vitro that were facilitated by PR's proline-rich domain (Ballare et al. 2003). However, this interaction was not sufficient to activate downstream components of the MAPK pathway (i.e. ERK2).
A putative common-docking (CD) domain has recently been identified in the N-terminal BUS unique to PR-B. MAPKs, such as ERK1/2, interact with their upstream activators, MAPK kinases (MKKs), such as MEK1, through CD domains (Rubinfeld et al. 1999, Tanoue et al. 2000). MEK1 binding to the MAPK CD domain may serve to anchor MAPK in the cytoplasm of unstimulated cells (Rubinfeld et al. 1999). CD domains are characterized by a cluster of negatively charged amino acids (DxxD/E) thought to interact with positively charged amino acids on the partner protein. MKKs, MAPK phosphatases (MKPs), and other associated downstream kinases contain positively charged ‘D’ domains (Tanoue et al. 2000, Ranganathan et al. 2006). CD domains, which are conserved throughout the MAPK family, contribute to the binding specificity of MAPKs with their respective MKKs. The putative CD domain within PR, DPSDE, exactly matches that of ERK2 and predicts direct PR binding to MEK1. We were able to detect endogenous PR/MEK1 interactions in T47D cells (Hagan et al. 2008). The functional significance of the PR CD domain is currently under investigation; PR/MEK1 complexes may stabilize, localize, and/or act to recruit MAPKs, in order to mediate post-translational phosphorylation events (i.e. at PR Ser294 and Ser345 MAPK consensus sites) required for nuclear PR actions. The interaction between PR/MEK1 may act as a scaffold to position MEK1 in close proximity to key components of the MAPK-signaling pathway (c-Src, EGFR, and ERK2) known to be rapidly activated by ligand-bound PR.
In summary, PRs contain multiple distinct domains (proline-rich, ERID-I and -II, and CD domain) that facilitate interactions with membrane-associated or cytoplasmic kinases, thereby modifying downstream signaling events. The significance of PR's role in extra-nuclear signaling, such as ERK1/2 activation, is supported by the presence of numerous protein kinase-interacting and scaffolding domains. PR's rapid signaling is fully integrated with its genomic actions as progesterone-activated protein kinases in turn directly phosphorylate PR and its coregulatory molecules leading to changes in gene regulation (Narayanan et al. 2005a, Daniel et al. 2007b, Faivre et al. 2008). Additionally, the PR DBD and the polyproline motif clearly contribute to the proliferative actions of progesterone (Faivre & Lange 2007).
PR interacts with cyclins, cyclin-dependent kinases, and cell-cycle inhibitors
Evidence from both animal and cell-line models suggests that PR signaling is tightly linked to cell-cycle regulation. This linkage may be therapeutically targeted in the clinic. ER and PR status have been positively associated with the overexpression of cyclins in breast tumors (Hui et al. 1996, Al-Kuraya et al. 2004, Reis-Fihlo et al. 2006, Millar et al. 2007). Striking similarities exist between cyclin D and PR knockout mice; both models exhibit identical phenotypes characterized by delayed lobuloalveolar development (Sicinski et al. 1995, Mulac-Jericevic et al. 2003). Similarly, the mammary glands of both cyclin D1 and PR-B transgenic animals exhibit hyperplasia (Wang et al. 1994, Shyamala et al. 2000). Several SRs interact directly or indirectly (via CDKs) with cyclins including ER (Zwijzen et al. 1998, McMahon et al. 1999), androgen receptors (AR; Knudsen et al. 1999, Reutens et al. 2001, Petre et al. 2002, Petre-Draviam et al. 2003, 2005, Burd et al. 2005, 2006), thyroid receptors (Lin et al. 2002), and PR (discussed below). Ongoing studies in our laboratory are examining the functional significance of PR interactions with cell-cycle regulatory molecules.
Acute exposure of cultured breast cancer cells to progestin rapidly upregulates cyclin D and initiates one or more rounds of cell-cycle progression, yet prolonged progesterone exposure subsequently induces growth arrest and insensitivity to further progesterone treatment. Progestin treatment is associated with upregulation of tyrosine kinase (erb B) growth factor receptors, in effect ‘priming’ cells for alternate mitogenic stimuli such as EGF or heregulin (Musgrove et al. 1993, Groshong et al. 1997, Lange et al. 1998, 1999, Labriola et al. 2003, Lange 2004). By contrast, Musgrove et al. (1998) reported that long-term exposure of breast cancer cell lines to progestins resulted in decreased overall cyclin D1, cyclin D3, and cyclin E expressions as well as the inhibition of cyclin D1/CDK4, cyclin D3/CDK4, and cyclin E/CDK2 complex kinase activities. Breast cancer cells, which are continuously exposed to progestin, show increased expression of the CDK inhibitors p21Cip1 and p27Kip1 (Groshong et al. 1997) and increased association of cyclin E/CDK2 complexes with p27Kip1 (Musgrove et al. 1998). Addition of exogenous cyclin D1 to progestin-inhibited cells reinitiates cell-cycle progression and causes the return of CDK2 activity (Musgrove et al. 1998). These data suggest that one function of cyclin D1 overexpression in breast cancer cells may be to provide a ‘sink’ for upregulated p27Kip1, thereby removing it from cyclin E/CDK2 complexes. Similarly, cells that continually overexpress cyclin D1 were not cell-cycle inhibited following long-term progestin treatment, yet showed increases in the proportion of cyclin E/CDK2 complexes associated with p27Kip1. These data suggest that cyclin D may also contribute to progestin-regulated cell-cycle progression by mechanisms that are independent of its ability to bind and sequester p27Kip1 (Musgrove et al. 2001). While cyclin E/CDK2 complex kinase activity is generally hindered by elevated p27Kip1, cyclin D/CDK4 complexes are sensitive to the CDK4/6 inhibitor p18INK4. Exposure of T47D cells to progestin increased p18INK4 expression leading to the inhibition of both cyclin E/CDK2 activity and cell-cycle progression (Swarbrick et al. 2000). Thus, chronic exposure of epithelial-derived breast cancer cells to progestin results in the upregulation of multiple CDK inhibitors that may initially nucleate and activate cyclin/CDK complexes (LaBaer et al. 1997), but ultimately decreases the activity of cyclin E/CDK2 and blocks cell-cycle progression (i.e. in the absence of other mitogenic stimuli). Overexpression of cyclin D, E, or A molecules or the loss of p21Cip1 or p27Kip1 in PR-positive breast cancer is predicted to bypass these cell-cycle controls (Musgrove et al. 1998). Additionally, these data are relevant to understanding the consequences of cyclical (i.e. as in menarche) versus chronic administration (i.e. as during post-menopausal hormone replacement therapy) of progestins; translation of these studies to the clinic must consider the complex biphasic actions of progesterone on breast epithelial cells.
CDK2-dependent regulation of PR Ser400
Differential phosphorylation of PR and the closely related glucocorticoid receptor alters transcriptional activity and is a mechanism for promoter selectivity (Daniel et al. 2007b, Blind & Garabedian 2008, Chen et al. 2008, Galliher-Beckley et al. 2008). Out of the 14 confirmed PR-B phosphorylation sites, 8 are cyclin/CDK2 targets (Moore et al. 2007). For example, one well-characterized CDK2 phosphorylation site on PR is Ser400, which is both basally phosphorylated and sensitive to mitogens (Pierson-Mullany & Lange 2004). Ser400 phosphorylation via CDK2 was required for rapid PR nuclear localization and robust ligand-independent PR activity in multiple p27-null cell-line models (Pierson-Mullany & Lange 2004). Additionally, both liganded and unliganded PR transcriptional activity, as measured using a progesterone response element (PRE)-luciferase reporter gene construct, increased as a result of forced expression of a constitutively active kinase mutant of CDK2 in breast cancer cells (Pierson-Mullany & Lange 2004). Nonetheless, the functional significance of PR/cyclin/CDK interaction is not fully understood. Not surprisingly, PR interacts with cyclin E/CDK2 and cyclin A/CDK2 complexes (Pierson-Mullany & Lange 2004, Narayanan et al. 2005a). Narayanan et al. (2005a) showed that overexpression of cyclin A increased ligand-induced PR transcriptional activity on a MMTV-luciferase reporter gene construct during S-phase in a CDK2-dependent fashion. The mechanism of increased PR transcriptional activity occurs via increased cyclin A/CDK2 phosphorylation of steroid receptor coactivator-1, which, in turn, displays increased association with PR.
Phosphorylation of PR MAPK sites mediates promoter selectivity
Serine 294
PR phosphorylation at Ser294 contributes to the regulation of hormone responsiveness, in part by directing promoter selectivity through a mechanism of PR sumoylation/desumoylation. Activation of the MAPK (ERK1/2) signaling pathway by growth factor receptor tyrosine kinases (e.g. via EGF; Qiu et al. 2003) or progestin-dependent PR/c-Src rapid signaling results in PR Ser294 phosphorylation (Shen et al. 2001, Skildum et al. 2005). Breast cancer cells (T47D) pretreated for 15–30 min with EGF prior to progestin exposure displayed heightened PR transcriptional activity relative to growth factor naive cells (Qiu & Lange 2003); cells expressing phospho-mutant S294A PR remained insensitive to EGF pretreatment (Qiu & Lange 2003, Daniel et al. 2007b). The insensitivity of mutant S294A PR to growth factors suggests that phosphorylation at Ser294 may induce PR hypersensitivity to low-progestin concentrations relative to non-phosphorylated receptors. Notably, S294A phospho-mutant PRs often exhibit impaired transcriptional responses when stably expressed (i.e. at levels comparable with endogenous PRs; Shen et al. 2001). However, decreased transcriptional activities are overcome when S294A PR is expressed at high concentrations (i.e. as in transient transfection assays; Shen et al. 2001, Qiu & Lange 2003). Thus, progestin and/or growth factor-induced phosphorylation on Ser294 may function to block/remove a repressive modification of PR in association with PRE-driven promoters, perhaps mediated by a limiting factor(s).
Related to these findings, transcriptional repression of target genes is often mediated by sumoylated transcription factors, and many transcription factors are modified by small ubiquitin-like modifier (SUMO) attachment in a phosphorylation-dependent manner (e.g. ELK-1, c-FOS, and AIB1; Yang et al. 2003, Bossis et al. 2005, Wu et al. 2006). SUMO (∼10 kDa) can be reversibly attached to lysine residues of target proteins thereby altering protein–protein interactions, subcellular localization, stability, and/or transcriptional activity (reviewed in Geiss-Friedlander & Melchior (2007)). Sumoylation, similar to ubiquitination, is a post-translational event requiring an enzymatic cascade in which SUMO molecules are processed and attached to target proteins via E1, E2 (UBC9), and E3 enzymes. The enzymes responsible for desumoylation, SENPs, are regulated in a hormone-dependent manner in prostate (Cheng et al. 2006) and mammary epithelial cells (Daniel et al. 2007a). A subset of PR is sumoylated at Lys388 in response to treatment with progestins (Abdel-Hafiz et al. 2002). The SUMO-deficient mutant receptor, K388R PR, displays tenfold increased transcriptional activity when expressed in breast cancer cells either transiently or stably (Abdel-Hafiz et al. 2002, Daniel et al. 2007a). Additionally, sub-physiological concentrations of progestin (10−11 M R5020) activated SUMO-deficient (20-fold) but not wild-type PR-B in breast cancer cells (Daniel et al. 2007a), demonstrating that K388R PR-B is transcriptionally hyperactive relative to the wild-type receptor. Alternatively, phospho-mutant S294A PR-B functions as a weak transcription factor and is more sumoylated relative to wt PR. These data suggest that sumoylation at PR Lys388 results in transcriptional repression, while phosphorylation at Ser294 reverses this effect. Indeed, Daniel et al. (2007a) have recently demonstrated that PR Ser294 phosphorylation negatively regulates sumoylation at PR Lys388.
PR-B sumoylation occurs in response to both progestin and anti-progestin (Chauchereau et al. 2003, Daniel et al. 2007a). EGF pretreatment of cells induced ERK1/2 activation, phosphorylation of Ser294, and PR-B desumoylation (Daniel et al. 2007a), whereas EGF-naive cells were not persistently phosphorylated at Ser294 and retained PR-B sumoylation in the presence of ligand. Forced phosphorylation at PR Ser294, via expression of constitutively active CDK2 (CDK2-TY) or MEK-1, blocked ligand-induced PR sumoylation, creating a hyperactive receptor; Ser294-dephosphorylated PR remained heavily sumoylated (Daniel et al. 2007a). These data indicate that PR sumoylation/desumoylation provides a phosphorylation-dependent mechanism for rapid derepression of PR transcriptional activity. PR Lys388 sumoylation shifts the progestin dose–response curve to the right, while Ser294 phosphorylation reverses this effect.
Sumoylated (S294A) PR are transcriptionally repressed relative to desumoylated (i.e. phosphorylated wild-type or K388R) receptors on selected endogenous promoters. Daniel et al. (2007a) showed HB-EGF, an endogenous PR target gene, was upregulated fivefold by SUMO-deficient K388R PR-B compared with wild-type following progestin treatment. Interestingly, IRS-1 expression was insensitive to progestin, but dependent upon PR-B Ser294 phosphorylation (Qiu & Lange 2003), and IRS-1 is upregulated in breast cancer cells stably expressing SUMO-deficient PR (unpublished observation). Other endogenous classical PR target genes, such as tissue factor (Kato et al. 2005), MUC1 (Brayman et al. 2006), and SGK (Jeong et al. 2005), are regulated similarly by both wild-type or sumo-deficient K388R PR-B (Daniel et al. (2007a) and unpublished observations). These data demonstrate that PR-B Ser294 phosphorylation or Lys388 sumoylation can dramatically affect transcriptional activation at a subset of endogenous PR-regulated promoters, yet have no effect on others. The underlying mechanism for PR gene selectivity remains unknown, but likely involves SUMO-dependent recognition of complex sequences (that may be distant) in association with PRE- or PRE half-site-containing promoter regions and their associated proteins (Holmstrom et al. 2003).
Serine 345
The mechanism governing SR regulation of the so-called ‘non-classical’ target genes lacking PREs is unknown. Of particular interest is how rapid signaling events may influence promoter selectivity. Recent work from Faivre et al. (2008) has uncovered a mechanism by which rapid progesterone/PR-initiated kinase signaling alters the phosphorylation state of PR resulting in the targeting of PR to SP1 sites in the promoter regions of endogenous genes. Following 10 min of progestin treatment, PR-B rapidly activated EGFR, c-Src, and MAPK signaling resulting in PR Ser345 phosphorylation in T47D cells (Faivre et al. 2008). PR Ser345 is a proline-directed MAPK consensus site located in the N-terminal region of the receptor. Phosphorylation of PR Ser345 is entirely ligand dependent (Fig. 2), yet completely blocked by inhibitors of EGFR, c-Src, and MAPK activity. To confirm that progestin-initiated rapid cytoplasmic signaling is required for PR Ser345 phosphorylation, the authors employed the mPro mutant PR lacking the polyproline motif required for PR interaction with c-Src (Boonyaratanakornkit et al. 2001). mPro PR-B fails to activate ERK1/2 MAPK and, in turn, these receptors do not undergo Ser345 phosphorylation. In the absence of progestins, EGF stimulation induced phosphorylation of a different PR MAPK consensus site, Ser294, but was unable to induce PR Ser345 phosphorylation indicating absolute specificity for progestin-initiated rapid signaling. Ligand-dependent PR/c-Src/MAPK complex formation and rapid signaling thus prime PR (i.e. via Ser345 phosphorylation) for downstream genomic actions (Figs 2 and 3).
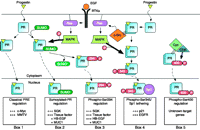
Post-translational modification of PR results in the regulation of specific subpopulations of PR genes. A subset of PR is sumoylated on Lys388 in response to progestin binding. Cells respond to growth factors (e.g. EGF) by activation of MAPK signaling resulting in PR phosphorylation at Ser294 and desumoylation at Lys388. These signaling mechanisms provide three different post-translationally modified populations of PR that can activate gene transcription at selected promoters (Boxes 1–3). Additionally, progestin treatment results in PR-mediated activation of c-Src through direct interaction of PR's proline-rich motif with c-Src's SH3 domain (Boonyaratanakornkit et al. 2001). Progestin-induced c-Src activation results in MAPK signaling and phosphorylation at PR Ser345 (Faivre et al. 2008). Phosphorylated PR at Ser345 can tether to Sp1 and activate transcription at promoters with Sp1-binding sites (Box 4). PR interactions with CDK2 result in increased liganded and unliganded PR activity on unknown PR target-gene promoters (Box 5; Pierson-Mullany & Lange 2004). Many additional genes are likely to be differentially regulated by post-translationally modified PR transcription factors.
Faivre et al. (2008) then used PR phospho-mutant S345A receptors to examine the consequences of PR Ser345 phosphorylation on PR target-gene regulation. S345A PR-B transcriptional activity was comparable with wild-type PR-B on classical PR promoters (2xPRE-luciferase reporter and the endogenous SGK promoter). Conversely, S345A PR-B was unable to activate transcription on non-classical progestin-responsive promoters such as p21 and EGFR, both of which contain numerous SP1 sites (Ishii et al. 1985, Hudson et al. 1990, Owen et al. 1998). Liganded wild-type PR-B, but not S345A or mPro, copurified with SP1 and was capable of tethering to SP1 sites in the p21 promoter as measured by chromatin immunoprecipitation assays. PR Ser345 phosphorylation in response to rapid signaling events therefore functions to target PR to specific promoters containing SP1 sites (i.e. p21 and EGFR) via PR/SP1 tethering (Faivre et al. 2008). PR induction of rapid c-Src/MAPK activation thus provides a concrete example of PR's ability to integrate rapid membrane-initiated signaling events with its genomic actions through a feed-forward mechanism (Fig. 3). PR-initiated rapid signaling is critical for progestin-induced S-phase entry in breast cancer cells (Skildum et al. 2005). Faivre et al. (2008) showed that blockade of EGFR, c-Src, MAPK, or SP1 via their respective inhibitors abolished progestin induced S-phase entry and anchorage-independent growth of T47D cells expressing wild-type PR-B. These data demonstrate that a specific PR phosphorylation event (Ser345) mediates PR selection of certain target genes directly responsible for the initiation of cell-cycle progression in breast cancer cells. Progestin-responsive genes regulated by PR tethering (i.e. via SP1, AP1 (Tseng et al. 2003), or STAT (Richer et al. 2002a,b, Proietti et al. 2005) molecules) may include a large portion of known PR target genes.
Conclusions
PRs are able to interact with and activate protein kinases functioning as part of signaling cascades resulting in subsequent modification of associated protein substrates, including PR itself. These early and rapid events ultimately direct PR to specific promoters via phosphorylation-dependent tethering interactions, leading to altered cell-cycle progression. Interaction of PRs with the components of MAPK modules and/or MAPK-regulated CDKs provides an exquisite sensing mechanism for PR function in the context of multiple hormonal inputs emanating from the cell surface (Fig. 3). We propose that differentially modified SR species shuttle to different promoters. Thus, experiments examining the transcriptional consequences of specific SR post-translational modifications must focus on the regulation of endogenous genes rather than deriving conclusions based solely on hormone response element-driven reporter constructs. We conclude that the cytoplasmic and genomic actions of SRs are often fully integrated in order to produce precisely timed events, such as cell-cycle entry in the case of PR. Targeting the dual activities of PR (and by analogy ER or AR) as both mediators of signal transduction and nuclear transcription factors using a combination of selective protein kinase inhibitors and anti-progestins (i.e. transcriptional antagonists) is likely to be a productive strategy for blockade of breast cancer progression.
Declaration of interest
We declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by National Institutes of Health Grants RO1 CA123763 and R21 CA116790 as well as an NIH University of Minnesota Cancer Center Postdoctoral Training Grant T32 CA009138 (to C R Hagan).
- © 2009 Society for Endocrinology










