The diagnosis and management of parasellar tumours of the pituitary
- Endocrine Unit, Department of Pathophysiology, National University of Athens, Athens, Greece1Department of Academic Radiology, St Bartholomew's Hospital, London, UK2Department of Endocrinology, Theagenion Hospital, Thessaloniki, Greece3Department of Endocrinology, St Bartholomew's Hospital, London EC1A 7BE, UK
- (Correspondence should be addressed to A B Grossman; Email: a.b.grossman{at}qmul.ac.uk)
Abstract
The sellar and parasellar region is an anatomically complex area where a number of neoplastic, inflammatory, infectious, developmental and vascular diseases can develop. Although most sellar lesions are due to pituitary adenomas, a number of other pathologies involving the parasellar region can present in a similar manner. The diagnosis of such lesions involves a multidisciplinary approach, and detailed endocrinological, ophthalmological, neuroimaging, neurological and finally histological studies are required. Correct diagnosis prior to any intervention is essential as the treatment of choice will be different for each disorder, particularly in the case of primary malignant parasellar tumours. The complexity of structures that define the parasellar region can produce a variety of neoplastic processes, the malignant potential of which relies on histological grading. In the majority of parasellar tumours, a multimodal therapeutic approach is frequently necessary including surgery, radiotherapy, primary or adjuvant medical treatment and replacement of apparent endocrine deficits. Disease-specific medical therapies are mandatory in order to prevent recurrence or further tumour growth. This is particularly important as neoplastic lesions of the parasellar region tend to recur after prolonged follow-up, even when optimally treated. Apart from the type of treatment, identification of clinical and radiological features that could predict patients with different prognosis seems necessary in order to identify high-risk patients. Due to their rarity, central registration of parasellar tumours is required in order to be able to provide evidence-based diagnostic and mainly therapeutic approaches.
Introduction
The area immediately around the pituitary, the sellar and parasellar region, is an anatomically complex area that represents a crucial crossroads for important adjacent structures (Ruscalleda 2005). While the sellar region has specific anatomical landmarks, the parasellar region is not clearly delineated and includes all the structures that surround the sella turcica (Rennert & Doerfler 2007). Vital structures such as the brain parenchyma, meninges, visual pathways and other cranial nerves, major blood vessels, hypothalamo-pituitary system (HPS) and bony compartments may be involved. A diversity of clinical symptoms and signs can develop from a number of neoplastic, inflammatory, infectious, developmental and vascular diseases that occupy the parasellar area secondary to the location, size and growth potential of the lesions, and the subsequent damage to specific adjacent vital structures (Freda & Post 1999).
Pituitary adenomas are the most common cause of a sellar mass extending to the parasellar region (Freda & Post 1999). However, in ∼9% of cases, an aetiology other than a pituitary adenoma is encountered, parasellar tumours being the second commonest cause after non-tumorous cystic lesions (Freda et al. 1996). The malignant potential of these tumours may be defined according to the World Health Organization (WHO) classification of tumours of the central nervous system (CNS), which relies on histological criteria: WHO grade I (tumours with low proliferative potential and the possibility of cure following surgical resection), WHO grade II (infiltrative tumours with low mitotic activity that can recur and progress to higher grades of malignancy), WHO grade III (tumours with histological evidence of malignancy) and WHO grade IV (mitotically active tumours with rapid evolution of disease; Kleihues et al. 1993). The latest version of the WHO classification, including some new histologically identified variants that may have some clinical relevance, has recently been published (Louis et al. 2007). A number of other non-neoplastic lesions, such as inflammatory, granulomatous, infectious and/or vascular pathologies can also involve the parasellar region (Table 1).
Classification of parasellar tumours according to malignant potential and other parasellar lesions
In this paper, we primarily attempt an overview of the clinical, endocrine and radiological features of neoplastic parasellar tumours, taking into consideration their malignant potential (Table 1). In addition, currently available therapeutic schemes and the long-term prognosis of these tumours will also be discussed. Non-neoplastic parasellar lesions will also be briefly mentioned but not discussed into detail.
Anatomical considerations
The parasellar region includes, laterally, the dural walls of the cavernous sinus, a multi-lobulated venous structure containing the intracavernous portion of the internal carotid artery, cranial nerves III, IV, VI and the V1 and V2 branches of the trigeminal nerve (Smith 2005; Fig. 1). The relationships of the cavernous sinus are: inferiorly with the basisphenoid and sphenoid sinus; superiorly, the diaphragma sella, with the suprasellar subarachnoid spaces containing the optic nerves and chiasm, hypothalamus, tuber cinereum and anterior third ventricle (Ruscalleda 2005). The nasopharynx and the medial aspects of the temporal lobes are also closely related to the parasellar region; in addition, embryonal remnants can also be found contributing further to the diversity of the processes that are involved in delineating the anatomy of this region (Smith 2005).
Clinical presentation
Non-pituitary adenomatous parasellar lesions do not present with hypersecretory syndromes but rather with hypopituitarism or symptoms of mass effect due to compression of nearby vital surrounding structures, the severity of which depends on the location, size and growth potential of the tumours (Glezer et al. 2008). Visual loss is a common presenting complaint due to the proximity of tumours to the optic nerves, chiasm and optic tracts. Headache develops either as a consequence of increased intracranial pressure (IP), distortion of the diaphragma or irritation of the parasellar dura (Freda & Post 1999). As parasellar tumours commonly originate or infiltrate structures within close proximity to the cranial nerves traversing through the cavernous sinus, cranial nerve abnormalities develop in ∼25% of cases (Jagannathan et al. 2007). Hypothalamic tumours in children may produce the diencephalic syndrome manifest as wasting, poor development and sexual immaturity, whereas in adults it may lead to disruption of the control of appetite and cause severe obesity or starvation (Freda & Post 1999). Involvement of the hypothalamus and pituitary leads to complete or partial pituitary hormonal deficiencies; hyperprolactinaemia may also develop secondary to stalk compression (Glezer et al. 2008). Diabetes insipidus (DI) is a common finding in parasellar tumours and indicates that the lesion is unlikely to be a pituitary adenoma; however, it cannot reliably distinguish among the various parasellar tumours (Freda & Post 1999).
Imaging
Radiological imaging of the parasellar region is challenging since the sella is a small volume region in close proximity to many complex structures (Ruscalleda 2005). Both thin-section computerised tomography (CT) and magnetic resonance imaging (MRI) play an important role in the anatomical delineation of lesions in this area (Boardman et al. 2008). MRI is the modality of choice providing multiplanar high-contrast images, whereas CT has a complementary role in delineating bony destruction and the visualisation of calcification (Rennert & Doerfler 2007). Conventional radiology is no longer in use, whereas digital subtraction angiography has been largely replaced by the continuous improvement in MR and CT angiographies (Rennert & Doerfler 2007). Although the radiological features of parasellar tumours have many similarities, some may have distinctive findings (Chong & Newton 1993, Freda & Post 1999, Ruscalleda 2005, Smith 2005; Table 2). However, the differential diagnosis among the various types of tumours remains difficult using currently available methods of morphological imaging.
Imaging characteristics of parasellar tumours
Diagnosis
In formulating a management plan, histological confirmation is usually required unless there are lesion-specific clinical, endocrine and/or radiological features. Existing methods of skull base biopsy in the anatomically critical parasellar region include either open skull base approaches or image-guided needle biopsy (Day 2003, Samandouras et al. 2005). Image-guided techniques may help in diagnosis, but can be time-consuming and cannot reliably avoid surrounding critical neurovascular structures; in selected patients endoscopic transnasal biopsy can be applied (Frighetto et al. 2003, Samandouras et al. 2005). However, the use of such methods should be weighed along with the potential morbidity and mortality of the procedures when the risk of inducing damage to a vital structure is high, while the expected benefit from obtaining the correct diagnosis may be questionable.
Therapy
Surgery remains the treatment of choice for benign tumours and malignant tumours that are not responsive to other forms of treatment, and/or when surgical debulking is essential (Day 2003, Glezer et al. 2008).
Surgical therapy
Parasellar tumours usually have irregular margins and adhere to vital neurovascular structures, and thus do not allow a complete resection without the danger of affecting critical brain areas (Couldwell et al. 2004). Resection is attempted by craniotomy and/or transsphenoidally (TSS), particularly for smaller tumours approachable through the sella (Baskin & Wilson 1986, Honegger et al. 1992). For massive lesions, a two-stage removal procedure may be necessary; initially TSS debulking followed by craniotomy later. This approach may allow residual tumour to descend inferiorly, facilitating further resection (Maira et al. 1995). Recently, the development of advanced cranial base TSS approaches has facilitated the exposure of basal lesions by the removal of osseous structures, minimising brain retraction and providing safe alternatives when assessing lesions involving the tuberculum sella, suprasellar region, cavernous sinus or clivus (Couldwell et al. 2004). The TSS route can also be used for midline lesions without significant lateral extension (Day 2003). In cases of hydrocephalus, resection may be achieved following decompression of the ventricles and stabilisation of the clinical status of the patient. In the presence of large cystic lesions, as with craniopharyngiomas, fluid aspiration can be applied first as may provide relief of the obstruction and facilitate further tumour removal (Karavitaki et al. 2006).
Radiotherapy (RT)
Conventional RT has traditionally been used either as primary treatment or as a way to prevent further tumour growth or recurrence (Brada & Cruickshank 1999, Perks et al. 1999). More sophisticated radiotherapeutic techniques have recently been introduced. Stereotactic radiosurgery delivers a single fraction of high-dose ionising radiation on mapped targets, keeping the exposure of adjuvant tissues to a minimum and allowing for the delivery of maximum tolerated dose of between 10 and 15 Gy (Giller & Berger 2005). Stereotactic RT combines the accurate focal delivery of stereotactic radiosurgery with the radiobiological advances of fractionation; compared with conventional RT, it minimises long-term toxicities by offering optimal sparing to surrounding tissue (Tarbell et al. 1994). Robotically controlled frameless radiosurgery has also been developed and studies assessing its efficacy are awaited (Giller et al. 2005). In general, RT seems to be a valuable asset in the treatment of these tumours, albeit with potential adverse effects to nearby tissue and the HPS (Brada & Cruickshank 1999).
Medical therapy
Chemotherapy remains the primary therapy for responsive malignant tumours in either a neoadjuvant or adjuvant setting following surgery and/or RT. Patients with infrequently curable or unresectable tumours should be considered candidates for clinical trials that evaluate interstitial brachytherapy, radiosensitisers, hyperthermia, or intraoperative radiation therapy in conjunction with external RT to improve local tumour control. Such patients are also candidates for studies that evaluate new drugs and biological response modifiers following RT. Therapy of established or evolving specific pituitary and hypothalamic hormonal deficits should be detected and adequately treated (Lamberts et al. 1998). Optimum hormonal replacement therapy should be aimed for, although even with replacement therapy parasellar tumours, particularly craniopharyngiomas, have a worse prognosis compared with pituitary adenomas (Tomlinson et al. 2001).
Follow-up
Prognosis is dependent on patient status, comorbid conditions, tumour size and extension, and the precise histopathology. Children and adolescents should have careful monitoring of height, weight and pubertal status, while a dedicated neuroendocrine team should screen patients for endocrine and neuropsychological deficits at regular intervals. Post-operative imaging should be performed within 3 months of treatment and generally at 6 and 12 months to evaluate tumour recurrence or presence of residual tumour.
Malignant tumours of the parasellar region
Gliomas
Gliomas may arise in the hypothalamus, optic chiasm, nerve or tract (Zee et al. 2003; Fig. 1). The main histopathological subtype is the pilocytic astrocytoma (WHO grade I), which is a low-grade malignant lesion associated occasionally with neurofibromatosis type 1 (NF1; Zee et al. 2003). In this instance, lesions can be multiple including optic nerve gliomas, low-grade brain stem gliomas and basal ganglia non-neoplastic hamartomas (Zee et al. 2003). The remainder of hypothalamic/optic chiasm lesions are diffuse astrocytomas (WHO grade II), representing 35% of all astrocytic brain tumours (Smith 2005). This type of tumour typically affects young adults and has a tendency for malignant progression to anaplastic astrocytoma and, very rarely, glioblastoma (WHO grades III–VI). Hypothalamic/chiasmatic astrocytomas are primarily seen in adulthood presenting with impaired vision and retro orbital pain (Black & Pikul 1999). In children, they may present with visual loss, headache, proptosis and the diencephalic syndrome (Glezer et al. 2008). On imaging, hypothalamic/chiasmal gliomas are usually large suprasellar masses, infiltrating the brain and third ventricle, which enhance homogeneously; rarely, there is necrosis, haemorrhage or calcification (FitzPatrick et al. 1999). There is uncertainty as to the optimum therapy, and many can be simply observed; obviously, visual loss will determine the need for surgery. The mean survival time after surgical intervention is between 6 and 8 years, with considerable individual variation (Kitange et al. 2003). Optic pathway gliomas generally behave benignly, with very slow growth; however, tumours around the chiasm/hypothalamus can be more aggressive, exhibiting a 50% 5-year survival (Kitange et al. 2003).
Gliomas can also develop in the brainstem and extend into the parasellar region (Packer 2000, Guillamo et al. 2001). Diffuse intrinsic low-grade glioma (WHO grade II) is the most prominent type, whereas purely malignant brainstem glioma (WHO grades III–VI) occurs in 31% of cases (Guillamo et al. 2001). The median survival of low-grade gliomas is 7.3 years whereas that of high-grade gliomas was 11.2 months (Guillamo et al. 2001). Stereotactic biopsy has been reported to provide the diagnosis with minimal morbidity (Packer 2000). Although glucocorticoids and irradiation may temporarily improve symptoms, there seems to be no long-term benefit (Recinos et al. 2007). Occasionally, mixed gliomas may occur, for which temozolomide, an oral derivative of dacarbazine, may be useful therapy. Clinical improvement has been noted in 51% of patients, with a radiological response rate of 31% (Hoang-Xuan et al. 2004).
Germ cell tumours (GCTs)
Primary intracranial GCTs are neoplasms most commonly present in the first two decades of life (Janmohamed et al. 2002; Fig. 2). Like other extragonadal GCTs, CNS variants develop around the midline, with 80% arising around the third ventricle, mostly in the region of the pineal gland, followed by the suprasellar compartment and anterior hypothalamic regions (Jennings et al. 1985). Synchronous GCTs at both these sites are found at ∼5–10% (Jennings et al. 1985). Germinomatous GCT (GGCT) are the most exquisitely radiosensitive, whereas non-GGCT (NGGCT), comprising choriocarcinoma, teratoma, embryonal sinus (yolk sac) tumour and embryonal carcinoma, have a poorer overall response to treatment and a worse prognosis (Allen et al. 1987, Balmaceda et al. 1996). Non-GCT can also contain germinomatous elements (Calaminus et al. 2005). Pineal GCT classically give rise to raised IP, hydrocephalus and Parinaud's syndrome, whereas suprasellar GCT typically present with cranial DI, hypopituitarism and visual disturbance, and may lead to dissemination via the CSF (Legido et al. 1989, Janmohamed et al. 2002). Diagnosis is established by histology but a subgroup can be diagnosed on the basis of elevation of specific tumour markers (Calaminus et al. 2005), or typical clinical and radiological features. Yolk sac tumours secrete α-fetoprotein and choriocarcinomas β-human chorionic gonadotrophin (β-hCG) that can be detected in the serum and/or CSF (Calaminus et al. 2005). Approximately 10% of germinomas may contain syncytiotrophoblastic elements and secrete β-hCG (Matsutani et al. 1997). Germinomas appear as large lesions typically slight hyperdense on CT that enhance after contrast medium administration (FitzPatrick et al. 1999). On MRI, they appear isointense to brain on T1-weighted images and isointense-slightly hyperintense on T2-weighted images (Rennert & Doerfler 2007).
Sagittal enhanced T1-weighted image of a germinoma with both a suprasellar and pineal enhancing mass lesions.
Although histological confirmation remains the ‘gold standard’ diagnostic method, the combination of tumour marker estimation and imaging modalities can be used in secretory GCT (Packer et al. 2000, Calaminus et al. 2005). This diagnostic tool is underscored by the fact that several patients with parasellar GCT have developed surgical procedure-related complications (Calaminus et al. 2005). It is therefore important to avoid aggressive surgical intervention and develop treatment planning on non-invasive modalities, and only if in doubt to proceed to a biopsy (Janmohamed et al. 2002, Calaminus et al. 2005). In cases of non-secretory GCT histological diagnosis is helpful, but again this may be associated with considerable morbidity (Packer et al. 2000). A biopsy can usually be obtained with using stereotactic neurosurgery or neuroendoscopy; occasionally, the small tissue fragments obtained can result in histological specimens that are not representative of the entire lesion (Packer et al. 2000).
Treatment of GCT involves irradiation to the tumour bed to obtain local control, craniospinal irradiation to cover leptomeningeal tumour spread, and chemotherapy to eliminate leptomeningeal and systemic tumour dissemination (Gobel et al. 2001). When a pre-operative diagnosis of a GCT has been obtained, then surgical exploration is not necessary as these tumours are highly sensitive to chemotherapy and RT (Gobel et al. 2001, Janmohamed et al. 2002). Platinum-based chemotherapy has proven to be highly effective in non-seminomatous GCT (Allen 1987, Balmaceda et al. 1996) A cumulative dose of cis-platin of over 300 mg/m2 and tumour bed irradiation dose of ∼50–54 Gy, together with craniospinal irradiation, has a synergistic effect and achieves a long-term relapse-free survival between 60 and 70% (Janmohamed et al. 2002, Calaminus et al. 2005). Neoadjuvant chemotherapy and delayed residual tumour resection may be more appropriate than primary tumour resection followed by the same chemotherapy (Janmohamed et al. 2002). The presence of CNS dissemination warrants more aggressive treatment, such as high-dose chemotherapy with stem-cell transplantation (Guillamo et al. 2001), or intraventicular treatment (Osuka et al. 2007). Two recent studies in children and adults have evaluated long-term treatment sequelae, revealing mainly impairment of endocrine function, and rare visual, hearing and neurological deficits (Legido et al. 1989). In general, our own approach has been to initiate treatment with chemotherapy in lesions that appear to be characteristic of germinomas, even in the absence of positive tumour markers, and then to re-image several weeks after initial chemotherapy; rapid shrinkage of the tumour will usually be confirmatory of the diagnosis, and the treatment completed.
Primary parasellar lymphomas
Primary CNS lymphomas account for less than 2% of all intracranial lesions (Fine & Mayer 1993; Fig. 3). By definition, these tumours are extranodal and arise primarily in the craniospinal axis, and are distinct from systemic lymphomas that secondarily metastasise to the CNS (Megan Ogilvie et al. 2005, Liu et al. 2007). Although initially described in immunocompromised patients, a significant increase in the incidence of such lesions has recently been documented in immunocompetent patients (Corn et al. 2000). Lymphomas involving the sellar/parasellar region are even rarer, representing less than 1% of all cases in a large cohort of patients who underwent TSS (Freda & Post 1999). Recently, the number of primary parasellar lymphomas has substantially increased, the majority being of B-cell origin and only a minority of T-cell origin; a case of a NK/T-cell lymphoma has also been reported (Liu et al. 2007). Approximately 18 patients with sellar/parasellar lymphomas have been described with a mean age at presentation of 55.5 years and male/female ratio of 13:5 (Fine & Mayer 1993, Liu et al. 2007). Endocrine abnormalities are relatively common, with 72% of patients exhibiting anterior, and 39% posterior, pituitary deficiencies respectively (Megan Ogilvie et al. 2005, Liu et al. 2007). In a recent review systemic symptoms, such as fever of unknown origin, were the presenting symptoms in 22% of patients, whereas the commonest presenting compressive symptoms were headache (56%), diplopia (39%) and visual field defects (28%) respectively (Liu et al. 2007). MRI may demonstrate enhancing parasellar masses with diffuse enlargement of the pituitary (94%), suprasellar extension (44%), cavernous sinus extension (39%) and stalk thickening (22%; Liu et al. 2007). Lymphomas usually appear iso- or hyperdense on CT scanning and isointense on T1-weighted and slightly hypointense on T2-weighted images and enhance homogeneously after gadolinium administration (Buhring et al. 2001). The diagnosis of primary lymphomas is established histologically and their treatment includes surgery, chemotherapy and RT according to established protocols (Megan Ogilvie et al. 2005, Glezer et al. 2008).
Metastases to the parasellar region
Metastases to the parasellar region are relatively rare, found in less than 1% of patients undergoing TSS for sellar/parasellar lesions (Komninos et al. 2004). However, autopsy series have revealed that metastases to the region comprise between 0.14 and 28.1% of brain metastases, the most common primaries being breast and lung cancer in women and men respectively; however, virtually any neoplasm can metastasise to the pituitary (Chiang et al. 1990, Sioutos et al. 1996, Megan Ogilvie et al. 2005). In a significant number of patients, the parasellar area and the pituitary gland may appear macroscopically normal besides the presence of metastases that remain clinically non-significant (Ito et al. 2001). Most cases are found in the sixth–seventh decade of life as part of a generalised metastatic spread, commonly associated with multiple, particularly osseous metastases (Chiang et al. 1990, Sioutos et al. 1996). However, they can occur in young patients, and very occasionally are the first manifestation of an occult cancer or the only site of metastasis (Sioutos et al. 1996). Symptoms of mass effect and cranial nerve palsies are the most frequent symptoms; when metastatic lesions involve the pituitary gland, hormonal deficiencies, particularly DI, develop (Sioutos et al. 1996, Komninos et al. 2004). Although clinically indistinguishable from other lesions involving the area, specific clinical settings may point to the diagnosis (Komninos et al. 2004). The rapidity of development of symptoms of mass effect, particularly ophthalmoplegia secondary to VI nerve involvement, in association with the sudden onset of DI in patients over 50 years, strongly suggests the presence of metastases irrespective of a history of malignancy (Ruelle et al. 1992, Sioutos et al. 1996, Morita et al. 1998). Although not specific, radiology can help identify parasellar metastases in the presence of other brain metastases and/or rapidly growing lesions; involvement of the infundibular recess favours a metastatic lesion (Morita et al. 1998, Komninos et al. 2004). In the absence of a primary and/or other metastatic lesions, confirmation of the diagnosis relies on histology (Chiang et al. 1990). Treatment is mainly palliative and depends on the symptoms and the extent of systemic disease (Morita et al. 1998). Due to the invasive, vascular and haemorrhagic nature of the lesions, surgical excision is not usually possible (Sioutos et al. 1996). However, surgical debulking may offer improvement of symptoms of mass effect; this should be followed by local radiation, systemic chemotherapy and adequate hormonal substitution. Radiosurgery is sometimes a useful alternative, particularly for previously irradiated regions, and for smaller lesions (diameter <30 mm). The overall prognosis is poor, with a median survival of less than 2 years (Laigle-Donadey et al. 2005).
Supratentorial primitive neuroectodermal tumour is an embryonal tumour (WHO grade IV) of the cerebrum or suprasellar region composed of undifferentiated or poorly differentiated neuroepithelial cells, which have the capacity for differentiation along neuronal, astrocytic, ependymal, muscular or melanocytic lines (Ohba et al. 2008). Synonyms include cerebral medulloblastoma, cerebral neuroblastoma, cerebral ganglioneuroblastoma and ‘blue tumour’. This rare tumour occurs mainly in children with an overall 5-year survival rate of 34% (Ohba et al. 2008).
Ependymoblastoma is a rare, malignant, embryonal brain tumour (WHO grade IV) that occurs in neonates and young children. Ependymoblastomas are often large and supratentorial and generally relate to the ventricles, though they can occur in the parasellar region (Matthay et al. 2003). These types of tumours grow rapidly, with craniospinal dissemination, and have a fatal outcome within 6–12 months of diagnosis (Matthay et al. 2003).
Potentially malignant parasellar lesions
Craniopharyngiomas
Craniopharyngiomas are rare epithelial tumours that arise along the pathway of the craniopharyngeal duct (Bunin et al. 1998; Figs 4 and 5). They account for 2–5% of all primary intracranial neoplasms and follow a bimodal age distribution with a peak incidence rate in children and adults between the ages of 5 and 14 and 50 and 74 years respectively (Bunin et al. 1998). The majority show a suprasellar component (94–95%), and may extend into the anterior, middle or posterior fossa (Petito et al. 1976). Tumours can be solid (1–16%), but most are cystic or mixed (84–99%); cysts may be multiloculated and contain liquid that has high content of membrane lipids and cytokeratins (Karavitaki et al. 2006). Despite their benign histological appearance (WHO grade I), their infiltrative tendency into critical parasellar structures and aggressive behaviour, even after apparently successful treatment, heralds a significant morbidity and mortality (Karavitaki et al. 2006). Cases of malignant transformation have also been described, for which the role of previous RT has not been discounted (Nelson et al. 1988). There are two primary histological subtypes: the adamantinous and the papillary, but transitional or mixed forms have also been recognised (Zhang et al. 2002). The adamantinous subtype is the most common and bears some similarity to the adamantinoma of the jaw (Karavitaki et al. 2006), accounting for the calcification and the development of teeth encountered particularly in children (Petito et al. 1976). Adamantinous craniopharyngiomas tend to adhere to the surrounding brain tissue, often making complete surgical resection impossible (Petito et al. 1976). The papillary variety, mostly found in adults, is well circumscribed and shows less infiltration to adjacent tissues (Karavitaki et al. 2006). Although, headaches, nausea/vomiting, papillo-oedema, cranial nerve palsies and hydrocephalus are more frequent in children, a large series found no differences when compared the presenting manifestations among children and adults (Karavitaki et al. 2006). Spontaneous rupture of cystic craniopharyngioma is rare, but when it occurs can cause chemical ventriculitis and meningitis (Zee et al. 2003). Endocrine dysfunction in children manifests as growth failure in 93% or delayed sexual development in ∼20% (Freda & Post 1999). Many adults present with a variety of anterior pituitary hormone deficiencies and 23% develop DI (Freda & Post 1999). CT is the ideal modality for the evaluation of the bony anatomy, identification of calcification and in distinguishing the solid and cystic components of the tumour (Pusey et al. 1987). Pre- and post-contrast enhanced images identify the cystic lesions as a non-enhancing areas of low attenuation; the solid component and the cystic capsule appear as contrast-enhancing areas (Pusey et al. 1987). MRI following contrast enhancement offers valuable topographic and structural analysis (Karavitaki et al. 2006; Table 2). Radical surgery may be successful in selected tumours; however, when surgical removal was substantiated with radiological confirmation, complete removal was accomplished in 18–84% of cases, clearly a wide range (Hald et al. 1994). Post-operative RT following either complete or incomplete tumour removal is associated with significantly decreased recurrence rate; RT alone provides 10-year recurrence rates between 0 and 23% (Rajan et al. 1997). Although the optimum total dose or fractionated protocols have not been established, it seems that recurrences are fewer with total doses above 54 Gy (Karavitaki et al. 2006). Recurrent tumours develop at a mean interval between 1 and 4.3 years, but recurrences as late as 26 years have been described (Coke et al. 1998); such tumours exhibit higher microvessel density values (Vidal et al. 2002), and chromosomal aberrations (Lefranc et al. 2003). Stereotactic radiosurgery is used for well-defined residual tumour tissue after surgery or for the treatment of small solid recurrent tumours, especially after failure of conventional RT (Suh & Gupta 2006). In large cystic portions, multimodality approaches with installation of radioisotopes or bleomycin may provide further benefits (Karavitaki et al. 2006). Recurring tumours and those that have undergone malignant transformation have been treated with systemic chemotherapy and interferon-α, albeit with short-lived results (Karavitaki et al. 2006). However, the treatment for aggressive tumours remains to be assessed by trials including large number of patients with adequate follow-up. In general, our own approach has been to attempt surgical removal, TSS where possible, but not to be radical where this may in any way compromise the hypothalamus; this would generally be followed by standard fractionated RT.
Sagittal unenhanced T1-weighted image of a craniopharyngioma. The mass is inseparable from the chiasm and hypothalamus and is predominantly of low T1 signal suggesting a cystic mass, although without a high lipid content.
Chordomas and chondrosarcomas
Approximately 10% of non-pituitary parasellar lesions are cartilaginous, originating from the primitive notochord in the skull base, chordomas being more frequent than chondrosarcomas (Allan et al. 2001; Fig. 6). These are slowly growing tumours presenting with visual complaints, mainly diplopia, whereas a third of patients complain of headache (Korten et al. 1998). Less common presentations include dizziness, tinnitus, facial sensory deficits, ataxia and hemiparesis. Both tumours are associated with extensive bone destruction (Meyers et al. 1992) but significant endocrinopathy is unusual, although anterior pituitary deficiencies may be encountered, particularly with chordomas (Allan et al. 2001). Following the establishment of clearly defined histopathological criteria, a distinction can be made as both tumours are positive for S-100 protein but, unlike chordomas, chondrosarcomas are negative for cytokeratin markers (CAM 5–2) and epithelial membrane antigens (Salisbury & Isaacson 1986). Intracranial chordomas are most frequently seen around the third decade and in ∼10–15% can be intradural (Allan et al. 2001). Chordomas can reach considerable size at the time of diagnosis and patients may develop neck pain and nasopharyngeal obstruction, and some tumours may progress to malignant transformation (Rennert & Doerfler 2007). These tumours can extend along the entire skull base causing destruction of the sella instead of the ballooning often seen in pituitary adenomas (Glezer et al. 2008). Bone destruction and calcification occur in 50% of cases and are better seen on CT; on MRI they appear heterogeneously hyperintense on T2-weighted images and show marked contrast enhancement (Rennert & Doerfler 2007).
Coronal enhanced T1-weighted image of a chondrosarcoma. This is an enhancing mass involving the left side of the sella and the cavernous sinus, but arising from the skull base just off midline.
Less than 200 cases of intracranial chondrosarcomas have been described (Korten et al. 1998). These malignant tumours (WHO grades II–III) occur more commonly in the axial part of the skeleton, representing less than 5% of skull base tumours with ∼75% arising in the parasellar region (Kunanandam & Gooding 1995). Chondrosarcomas arise off the midline at the suture line, unlike chordomas that arise from the clivus in the midline. Radiological examination almost always reveals bone destruction and variable degrees of calcification on CT, involvement of the neural and vascular structures on MRI, and mostly hypovascularity on angiography (Korten et al. 1998). Occasionally, cyst-like hypodense centres secondary to necrosis can be found (Cohen-Gadol et al. 2003). Although chondrosarcomas rarely metastasise outside the skull they tend to have a better 5-year survival rate compared with chordomas, they expand locally and compress adjacent structures (Rosenberg et al. 1994). Surgery is the treatment of choice for both tumours; however, due to bone invasiveness, total excision is generally not possible (Glezer et al. 2008). Given the importance of residual tumour volume as a prognostic indicator, adjacent therapy is frequently employed (Allan et al. 2001). Standard external RT has been disappointing in achieving local control although most of the reported cases refer to chordomas, but radiosurgery may be more effective (Allan et al. 2001). Although a sustained response to the combination of ifosfamide and doxorubicin in a case of recurrent chondrosarcoma has been described, in most cases chemotherapy fails (La Rocca et al. 1999).
Haemangiopericytomas
Haemangiopericytomas are rare tumours accounting for less than 1% of all intracranial tumours (Glezer et al. 2008, Jalali et al. 2008). Although the majority are supratentorial, parasagittal or falcine, they can rarely arise in the sellar/parasellar region with less than ten cases reported (Jalali et al. 2008). These tumours are aggressive and highly vascular with numerous penetrating vessels; although previously considered as a subtype of angioblastic meningioma, they represent distinctive mesenchymal neoplasms arising from pericytes (Stout & Murray 1942). Besides the absence of firmly established histological criteria for grading haemangiopericytomas, they appear to correspond to WHO grades II–III (Kleihues et al. 1993). Most patients present in middle aged with visual field defects, headaches and rarely symptoms of endocrine dysfunction; a case of acromegaly due to a GHRH-secreting haemangiopericytoma has been described (Yokota et al. 1985). Surgery followed by RT is the standard treatment; however, as haemapericytomas are highly vascular and tend to bleed profusely, it is critical that the neurosurgeon bases his approach on a reasonable pre-operative diagnosis (Mena et al. 1991, Suh & Gupta 2006). Local tumour recurrence following treatment is common, and late widespread metastasis can occur (Jalali et al. 2008). In two large series haemangiopericytomas recurred in 91 and 85% after 15 years (Mena et al. 1991, Jalali et al. 2008). Cystic and necrotic areas, haemorrhage and prominent vascular channels contribute to the heterogeneity in signal intensity (Zee et al. 2003).
Langerhans' cell histiocytosis (LCH)
LCH is a rare disease characterised by the clonal accumulation and/or proliferation of specific dendritic cells and in this manner represents a neoplastic disorder (Arceci 1999; Fig. 7). LCH shows a particular predilection for involvement of the HPS leading to DI and/or anterior pituitary dysfunction (Arceci 1999, Makras et al. 2007), but can virtually involve any organ such as bone, lung, skin, liver, spleen, lymph nodes and bone marrow (Makras et al. 2007). Making an early and accurate diagnosis is important as multisystem LCH is associated with a 20% mortality rate, and 50% of those who survive develop at least one permanent consequence (Makras et al. 2007). Although there is no specific MRI appearance of HPS–LCH, almost all patients with LCH-induced DI demonstrate loss of the physiologic intense signal ‘bright spot’ of the posterior pituitary; other common MRI findings are infundibular enlargement, and/or the presence of hypothalamic mass lesions (Kaltsas et al. 2000). In cases where a precise diagnosis cannot be obtained histological diagnosis from the parasellar pathology may be required (Kaltsas et al. 2000). The combination of vinblastine with steroids is the most frequently used initial therapy of LCH involving the CNS and for multisystem disease (Arico et al. 2003). Although etoposide was initially extensively used, it is not used any long due to its associated high risk for the development of leukaemia (Arico et al. 2003). The purine analogue cladribine (2-chlorodeoxyadenosine, 2-CdA) has been shown to be effective for patients with recurrent and/or disseminated disease (Makras et al. 2007). RT, at doses up to 25 Gy, has been used in patients with endocrine deficiencies and radiological involvement of the parasellar region with partial or temporary radiological improvement (Arico et al. 2003).
Mainly benign lesions
Meningiomas
Meningiomas (WHO grade I) are the most common non-glial primary brain tumour comprising 15% of primary brain tumours with a peak incidence between 40 and 70 years (Zee et al. 2003; Figs 8 and 9). Tumours arise from the diaphragma sella, tuberculum sella, medial lesser wing of sphenoid, anterior clinoid, clivus cavernous sinus or optic nerve sheath (Smith 2005). In ∼10–15% of cases, meningiomas arise from the parasellar region and represent the commonest tumour of that region after pituitary adenomas; rarely, they occur entirely within the sella mimicking a pituitary adenoma (FitzPatrick et al. 1999). There is a strong association between meningiomas, NF (multiple tumours) and ionising radiation, depending on the dose applied (Hartmann et al. 2006). The clinical presentation is usually with visual disturbance and occasionally endocrine dysfunction with mildly raised prolactin levels (Freda & Post 1999). The visual field defects vary and depend on the location of the tumour; meningiomas may increase in size during pregnancy and then become symptomatic (Freda & Post 1999). Atypical (WHO grade II) meningiomas may develop in 4–7%, whereas anaplastic tumours (WHO grade III) are seen in 1–2.8% of cases (Hartmann et al. 2006). However, malignant behaviour, although very rare, may occur with any grade of meningioma (Bruna et al. 2007, Ko et al. 2007). Imaging features of meningiomas frequently allow pre-operative diagnosis, distinguishing them from other parasellar tumours (Smith 2005). Lesions arise from the dura and have a broad attachment to the dura or fill and expand the cavernous sinus. There may be a linear, enhancing dural tail extending along the dura away from the lesion. Bone reaction, with bone thickening and sclerosis, or expansion of the sphenoid sinus air space, may also be seen (Smith 2005). Meningiomas typically are similar to grey matter in CT density and T1- and T2-weighted MR image signal intensity, enhancing homogeneously and brightly with occasional areas of diffuse calcification (Smith 2005). In contrast to other parasellar tumours, meningiomas encase blood vessels and tend to narrow the lumen (Young et al. 1988). Surgical excision, particularly in the presence of symptomatic or growing lesions, remains the treatment of choice (WHO I). Following surgical excision, grade I tumours recur in 7–20%, whereas atypical and anaplastic recur in 39–40% and 50–78% respectively (Bruna et al. 2007, Ko et al. 2007, Nakane et al. 2007). Surgery and RT is used in selected cases, such as for patients with known or suspected residual disease or with recurrence after previous surgery, whereas radiation alone is used in patients with unresectable tumours (Freda & Post 1999, Aghi & Barker 2006). The prognosis is worse for patients with WHO grades II and III tumours as complete resection is less common and the proliferative capacity is greater (Bruna et al. 2007, Ko et al. 2007, Nakane et al. 2007). Malignant histology is generally associated with a poor prognosis with a survival less than 2 years (Hartmann et al. 2006, Bruna et al. 2007, Nakane et al. 2007).
Sagittal enhanced T1-weighted image of a suprasellar meningioma. The mass lies above the pituitary (which is normal), involves the chiasm and extends anteriorly along the planum sphenoidale.
Rathke's cysts
Rathke's cleft cysts arise from remnants of squamous epithelium of Rathke's pouch and consist of a single layer of epithelial cells with mucoid, cellular or serous components in the cyst fluid (Freda & Post 1999; Fig. 10). Although the majority has a major intrasellar component, about a third can extend into the parasellar region; occasionally they can be entirely suprasellar (Mukherjee et al. 1997, Freda & Post 1999). Many of these are small and asymptomatic, but they may become symptomatic due to compressive symptoms and pituitary hormonal deficiencies, when DI may develop in up to 20% (Mukherjee et al. 1997). Atypical presentations include haemorrhages into the cyst and abscess formation rarely can coexist with pituitary adenomas (Mukherjee et al. 1997). They are distinguished from craniopharyngiomas histologically by having walls composed of columnar or cuboidal epithelium (FitzPatrick et al. 1999). Although no specific radiological features have been identified, Rathke's cysts appear as discrete cystic and non-enhancing lesions on MRI with variable signal intensity on either T1- or T2-weighted images; these lesions need to be differentiated from craniopharyngiomas and arachnoid cysts (Mukherjee et al. 1997, Freda & Post 1999). Treatment involves drainage of the fluid with or without resection of the cystic wall (Mukherjee et al. 1997, Freda & Post 1999). Although these lesions tend not to recur, repeated resections may be required in as many as a third of patients; it has been suggested that many relapsing lesions may contain overlapping histological features with craniopharyngiomas (Mukherjee et al. 1997, Freda & Post 1999). The role of RT for recurrent lesions has not been clearly defined (Mukherjee et al. 1997).
Epidermoids/dermoids
These benign tumours (WHO grade I) comprise less than 2% of all intracranial tumours and arise as a result of incomplete separation of the neuroectoderm from cutaneous ectoderm (FitzPatrick et al. 1999). Epidermoids/dermoids occur in the cerebellopontine angle, pineal region, middle cranial fossa as well as in the suprasellar region, and consist of tissue of purely epidermal origin; dermoids also contain tissue of mesodermal origin (hair follicles, fat, sebaceous glands). Tumours are cystic structures lined by keratinised squamous epithelium and the clinical presentation may be due to local mass effect such as hydrocephalus, visual disturbance, hypopituitarism, DI and/or cranial nerve abnormalities (Freda & Post 1999). On CT scan, the cyst contents are very similar to CSF; both lesions are often hypointense on T1- and hyperintense on T2-weighted MR images and do not enhance with contrast. Depending upon fat and calcium content, dermoid tumours can show a hyperintense signal in T1 images (Rennert & Doerfler 2007). Diffusion-weighted images can be helpful in epidermoids, typically showing a markedly increased signal (Rennert & Doerfler 2007).
Hypothalamic hamartomas
Hypothalamic hamartomas are of neuronal origin and represent congenital heterotopias usually located within the tuber cinereum and mostly affecting children causing precocious puberty and epileptic seizures (Judge et al. 1977; Fig. 11). As they are usually less than 2 cm in diameter, they produce few symptoms of mass effect (Freeman et al. 2004). Typically, they appear as non-enhancing lesions and demonstrate rounded expansion of the tuber cinereum, best seen in coronal and sagittal images; they are isointense to the cerebral cortex in both T1- and T2-weighted MR images (Rennert & Doerfler 2007). The hamartomas associated with precocious puberty may contain GNRH1 neurons, and have been classically associated with ‘gelastic’ (laughing) seizures.
Paragangliomas
Paragangliomas are rare, usually encapsulated and benign neoplasms (WHO grade I), that arise in specialised neural crest cells associated with segmental or collateral autonomic ganglia (Freeman et al. 2004). Although the majority develop in the carotid body and jugular glomus, paraganglionic cells have been demonstrated in the pituitary gland and adjacent structures (Steel et al. 1993). Only seven cases of intrasellar, two cases of suprasellar and four cases of parasellar non-malignant paragangliomas have been described (Boari et al. 2006). Tumour location is more relevant than histology in assessing prognosis; the metastatic rate in para-aortic paragangliomas ranges between 28 and 42%, whereas only 5% of CNS paragangliomas exhibit metastatic potential (Boari et al. 2006). Symptoms of pituitary hormonal deficiency and mass effects are the main presenting symptoms (Steel et al. 1993, Boari et al. 2006). Surgical excision, when possible, remains the treatment of choice; there are no data regarding the prophylactic effect of RT on tumour recurrence.
Lipoma
Lipomas are benign fatty tumours, derived from remnants of maldevelopment of the primitive meninx (Smith 2005; Fig. 12). In the sellar region, they occur as lesions adherent to the surface of the infundibulum, floor of the third ventricle or adjacent cranial nerves (Smith 2005). They are usually discovered incidentally but rarely may enlarge and produce symptoms. Lipomas are all well-circumscribed, homogeneous lesions that appear identical to fat on CT and all MRI sequences, do not enhance and usually exhibit rim calcification (Smith 2005).
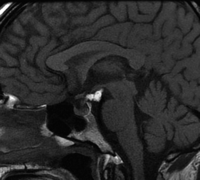
Sagittal unenhanced T1-weighted image of a suprasellar lipoma. The high signal mass containing fat is seen lying on the undersurface of the hypothalamus in the interpeduncular cistern.
Schwannoma/Neurinoma
Nerve sheath tumours (WHO grade I) in the parasellar region are very rare and usually arise from the trigeminal nerve (V1/V2) or the III, IV and VI cranial nerves (Rennert & Doerfler 2007; Fig. 13). Neurinomas are slowly growing tumours, rarely causing bony remodelling of the lateral portion of the sella or the apex of the petrous bone (Rennert & Doerfler 2007). Schwannomas are slow-growing tumours that may erode the walls of cavernous sinus, are found in association with NF2 and exhibit inactivating mutations of the NF2 gene in 60% (Aghi & Barker 2006). They usually develop symptoms of trigeminal nerve involvement and extremely rarely they can undergo malignant change (Aghi & Barker 2006). On MRI and CT, they show an intense (usually heterogeneous) contrast enhancement (Rennert & Doerfler 2007).
Parasellar granular cell tumours
These are rare tumours originating from either the neurohypophysis or infundibulum and include myoblastomas, choristomas and infundibulomas (Cone et al. 1990, Cohen-Gadol et al. 2003; Fig. 14). In 50% of cases cause anterior pituitary hormone deficiencies and hyperprolactinaemia, and rarely visual field defects (Freda & Post 1999). Although choristomas arise from the infundibulum and/or posterior lobe, only two cases of DI have been described; a case presenting with acromegaly secondary to GHRH secretion has also been described (Cohen-Gadol et al. 2003). These lesions are typically isointense to brain, and enhance inhomogeneously (Freda & Post 1999). Their firm and vascular nature, along with the lack of an obvious dissection plane between the tumour and normal brain, prohibits gross total resection (Cohen-Gadol et al. 2003). Although radiation therapy has been used, the recurrence rate in those who underwent RT was not different compared with those who did not, 4.7 and 5.4 years respectively (Cohen-Gadol et al. 2003).
Ganglion cell tumours (gangliocytoma)
These are rare tumours that consist of purely neuronal or mixed adenomatous and neuronal tissue, usually found in association with a hormonally active pituitary tumour (Freda & Post 1999). Gangliocytoma (WHO grade I) and ganglioglioma (WHO grades I–II) are well-differentiated, slow-growing neuroepithelial tumours comprised of neoplastic, mature ganglion cells, either alone (gangliocytoma) or in combination with neoplastic glial cells (ganglioglioma). Anaplastic gangliogliomas (WHO grade III) are sometimes seen; rare cases exhibit WHO grade IV (glioblastoma) changes in the glial component. The correlation of anaplasia with clinical outcome is inconsistent (Day 2003, Zee et al. 2003).
Rare parasellar tumours
Ependymomas are glial neoplasms that very rarely can develop in the parasellar region with only four cases described up to date; surgery with or without RT is the treatment of choice (Karim et al. 2006). Pituitocytoma is a primary tumour of the neurohypophysis, also located at the pituitary stalk, presenting with headache and hypopituitarism. Although histologically benign, the location and vascular nature of the tumour makes surgical resection difficult (Glezer et al. 2008). Other tumours such as plasmatocytomas, brown cell tumours and melanocytic tumours have been described as cases reports (Glezer et al. 2008).
Non-neoplastic pathologies involving the parasellar region
A number of non-neoplastic processes can also involve the parasellar region and present in a similar manner to parasellar neoplasms (Table 1). Their diagnosis is usually established in the presence of relevant clinical settings and if necessary histologically. Abscesses in the region can develop following direct extension from the sphenoid sinus, cavernous sinus and CSF or secondary to bacteraemia; masses of the sella can become secondarily infected (Freda et al. 1996). Tuberculosis can produce basilar meningitis and findings suggestive of a sellar/parasellar mass with hypopituitarism; there is usually evidence of tuberculosis elsewhere (Freda et al. 1996). Fungal, parasitic and opportunistic infections may also develop particularly in immunocompromised patients (Freda & Post 1999). Sarcoidosis can also present in a similar manner with varying degrees of hypopituitarism, DI and cranial neuropathy that in a minority can occur without evidence of sarcoidosis elsewhere (FitzPatrick et al. 1999). Other granulomatous diseases that can involve the parasellar region include giant cell granulomatous hypophysitis, Wegener's granulomatosis and the Tolosa–Hunt syndrome (Glezer et al. 2008). Aneurysms originating from the cavernous sinus or circle of Willis can project into the parasellar region and their appearance is mostly affected by the amount of calcification and thrombosis present within the aneurysms (Zee et al. 2003).
Summary
The parasellar region can be affected by a variety of tumours. The presentation of these tumours can be similar to that of large non-functioning pituitary tumours with extensive extrasellar extension and other non-neoplastic lesions involving the same region. Although symptoms of mass effect to adjacent structures and anterior pituitary endocrine deficits do not distinguish parasellar tumours from non-functioning pituitary tumours, the presence of DI directs towards a non-pituitary aetiology. Considering the specific morphological characteristics of MRI and CT, a presumptive diagnosis is possible in many cases of parasellar lesions. However, in the majority of cases their diagnosis involves a multidisciplinary effort including detailed endocrinological, ophthalmological, neurological and imaging procedures. In cases of doubt, a histological diagnosis may still be required to allow appropriate treatment planning. Treatment involves a joint effort requiring the collaboration of different specialties. Although a significant number of parasellar tumours are slow growing and mostly benign, it is important to identify those which are malignant and/or exert a strong malignant potential. Surgical decompression and/or clearance of the tumour along with means to prevent recurrence and/or further tumour growth, such as RT, are usually applied. Specific hormonal replacement therapy of established or evolving endocrine deficits is required to maintain the patient's quality of life. Primary or adjuvant medical treatment may be necessary for malignant, recurrent tumours and/or tumours undergoing malignant transformation. Multidisciplinary teams for the management and support of patients with these tumours are required for better long-term results. Tumour-specific therapies are required and need to be addressed on the basis of tumoral biology. The central registration of patients seems necessary as it may provide correlates between forms of treatments and outcomes and establish prognostic factors at the pathological or molecular level.
Declaration of interest
These authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
- © 2008 Society for Endocrinology










