Androgen receptor and growth factor signaling cross-talk in prostate cancer cells
- Departments of Urology and Toxicology, University of Kentucky College of Medicine, University of Kentucky Medical Center, Combs Research Building Room 306, Lexington, Kentucky 40536, USA
- (Correspondence should be addressed to N Kyprianou; Email: nkypr2{at}uky.edu)
Abstract
Androgens promote the growth and differentiation of prostate cells through ligand activation of the androgen receptor (AR). Sensitization of the androgenic response by multifunctional growth factor signaling pathways is one of the mechanisms via which AR contributes to the emergence of androgen-independent prostate tumors. The ability of AR to cross-talk with key growth factor signaling events toward the regulation of cell cycle, apoptosis, and differentiation outcomes in prostate cancer cells is established. In this paper, we review the functional interaction between AR and an array of growth factor signal transduction events (including epidermal growth factor; fibroblast growth factor; IGF1; vascular endothelial growth factor; transforming growth factor-β) in prostate tumors. The significance of this derailed cross-talk between androgens and key signaling networks in prostate cancer progression and its value as a therapeutic forum targeting androgen-independent metastatic prostate cancer is discussed.
Introduction
Prostate cancer development and growth is dependent on androgens and can be suppressed by androgen ablation monotherapy. Due to the emergence of androgen-independent prostate tumor growth however, prostate cancer recurs as androgen-independent, highly metastatic advanced disease (Wang et al. 2007).
Androgen functions through an axis involving testicular synthesis of testosterone, conversion by 5 reductase to the active metabolite 5 dihydrotestosterone (DHT), and its binding to androgen receptor (AR) to induce transcriptional activation of target genes (Siiteri & Wilson 1974, Imperato-McGinley et al. 1985, Heinlein & Chang 2002). In the adult prostate, androgens promote survival of epithelial cells, the primary step to malignant transformation to prostate adenocarcinoma (De Marzo et al. 1998). Androgen-induced prostate epithelial cell proliferation is regulated by an indirect pathway involving paracrine mediators produced by stromal cells, such as insulin-like growth factor (IGF), fibroblast growth factor (FGF), and epidermal growth factor (EGF; Cunha & Donjacour 1989, Byrne et al. 1996). The absence of a link between elevated serum testosterone, DHT, or adrenal androgens and prostate cancer risk suggests that androgens are not sufficient to promote prostate carcinogenesis (Roberts & Essenhigh 1986, Hsing 2001). The current evidence on the cross-talk between AR/androgen axis and signaling effectors of growth factors, as the contributing mechanism to prostate tumor initiation and progression, is discussed in this review.
AR connects with EGF
EGF and its membrane receptor, the epidermal growth factor-1 receptor (EGFR), are involved in the pathogenesis of different tumors, including prostate cancer (Russell et al. 1998). Both the ligand and its signaling receptor partner are frequently up-regulated in advanced stages of prostate cancer (Di Lorenzo et al. 2002). Targeting EGFR with monoclonal antibodies or with tyrosine kinase inhibitors suppresses growth and invasion of androgen-dependent and -independent prostate cancer cells in vitro (Bonaccorsi et al. 2004b, Festuccia et al. 2005). The involvement of EGFR in proliferation and invasion of cancer cells have been supported by other evidence (Wells et al. 2002). EGFR also participates in the formation of plasma membrane structures (lamellipodia) that mediate migration through the basal membrane (Rabinovitz et al. 2001). Significantly, elevated EGFR enhances the invasion potential of mammary tumors by increasing cell motility, without affecting tumor growth (Xue et al. 2006), pointing the key role exerted by the EGF/EGFR system in invasion and metastasis. Moreover, the robust evidence on the interaction between EGF/EGFR and androgen signaling provides proof of principle that engagement of multi-crossed signals is crucial for the acquisition and the maintenance of androgen sensitivity (Leotoing et al. 2007). Expression of the androgen-regulated prostate specific antigen, (KLK3) gene, is induced by the administration of interleukin-6 (IL6), which activates EGFR (Hobisch et al. 1998, Ueda et al. 2002). This evidence initially pointed to the contribution of EGFR in dictating AR outcomes in prostate cancer cells. ERBB2, a lead member of the EGFR family of receptor tyrosine kinases, was shown to be overexpressed in prostate cancer during progression to androgen-independent metastatic disease (Heinlein & Chang 2004). The mechanistic basis for important correlative cross-tall between AR and Erb2 has been provided by other reports indicating that modulation of AR signaling activity by the HER-2/neu tyrosine kinase promotes androgen-independent prostate tumor growth in vitro and in vivo (Craft et al. 1999, Yeh et al. 1999). More recent evidence further supports the signaling interaction by indicating that the loss of ERBB2 by siRNA impaired prostate cancer cell growth via targeting AR activity (Mellinghoff et al. 2004). Taken together, these lines of evidence converge to the recognition of the ERBB2 kinase activity being required for optimal transcriptional activity of AR in prostate cancer cells (Mellinghoff et al. 2004, Liu et al. 2005).
Androgens can post-transcriptionally control protein expression by regulating the binding of endogenous HuR to the AU-rich 3′UTRs, e.g. EGF mRNA (Myers et al. 1999, Torring et al. 2003). The ability of androgens to regulate the expression of androgen response element (ARE)-binding proteins that bind to these instability elements, supports an additional mechanistic involvement (by androgens) in the post-transcriptional control of EGF (Simons & Toomre 2000, DiNitto et al. 2003, Kuhajda 2006). In a ‘reversal-of-action’ mode, EGF reduces AR expression and blocks androgen-dependent transcription in differentiated cells, while it activates the AR promoter (Culig et al. 1994). This mechanistic EGF–AR interplay is an important contributor to prostate tumor progression, but it is not exclusive to EGF, as AR activity can be modulated by other growth factors (Orio et al. 2002).
AR interacts with the mitogen-activated protein kinase (MAPK)/extracellular signaling-regulated kinase kinase-1 (MEKK1) and the EGFR (Abreu-Martin et al. 1999, Bonaccorsi et al. 2004a; Fig. 1). Androgen-activated AR activates MAPK (Peterziel et al. 1999) and in a ‘functional-symmetry’, EGF-activated MAPK signaling cascade interferes with AR function, modulating the androgen response. MAPK extracellular kinase (MEK) inhibition reverses the EGF-mediated AR down-regulation in differentiated cells, thus suggesting the existence of an inverse correlation between EGF and androgen signaling in non-tumor epithelial cells (Leotoing et al. 2007). Additional key signal transducers in this dynamic, include transducer activator of transcription 3 (STAT3), most probably required for AR activation by IL6 toward promoting metastatic progression of prostate cancer (Abdulghani et al. 2008). Increased levels of Stat3 have been shown to lead to Stat3–AR complex formation in response to EGF and IL6 (as shown on Fig. 1). Moreover, Stat3 increases the EGF-induced transcriptional activation of AR, while androgen pre-treatment increases Stat3 levels in an IL6 autocrine-/paracrine-dependent manner suggesting an intracellular feedback loop (Aaronson et al. 2007). AR can also affect clathrin-mediated endocytosis pathway of EGFR, an essential step in its signaling integrity. The significance of engaging such an robust cross-signaling by prostate cancer cells toward determining their survival and response to the microenvironment is established by growing evidence (Bonaccorsi et al. 2007).
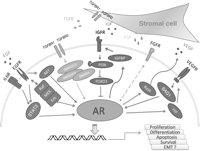
Growth factors cross-talk with AR in prostate cancer cells. IGF, FGF, VEGF, and TGFB secreted by the prostate stromal cells activate their receptors and interact with AR signal axis. In prostate epithelial cells, the androgenic signal engages secreted VEGF and TGFB which affects the prostate tumor microenvironment by inducing angiogenesis and stromal cell growth and differentiation. EGF signaling encounters AR signal in a tight control of multiple pathways. Growth factor signaling may proceed via AR signal and regulate the downstream effectors of AR regulating key cellular processes including proliferation, differentiation, apoptosis, and survival of prostate cancer cells.
The recently identified active integration of AR and EGFR signaling within the lipid raft microdomains in target cells provides an intriguing topological twist to this cross-talk. Thus, considering that the serine–threonine kinase AKT1 is a convergence point of the two hormonal stimuli and AR is localized in lipid raft membranes where it is stabilized by androgens (Freeman et al. 2007), one could easily argue that the newly found membrane ‘domain’ harboring AR is responsible for the non-genomic signaling by AR. The emerging concept that AKT1 is sensitive to manipulations in cholesterol levels, gains direct support from biochemical analysis verifying that a subpopulation of AKT1 molecules resides within lipid raft microdomains (Bauer et al. 2003, Zhuang et al. 2005). Distinct changes in phosphorylation state of AKT1 in response to androgen occur quickly but temporally independent in the raft and non-raft compartment, implicating processing of dissimilar signals. Interestingly, EGF triggers AKT1 phosphorylation via more rapid kinetics than those induced by androgens; this was recently documented by studies on the sensitivity of EGFR family proteins to disruptions in cholesterol synthesis and homeostasis, supporting the functional significance of EGF signal transduction through lipid rafts (Freeman et al. 2007).
AR and IGF interactions
Signaling by IGF1 is of major mechanistic and biological significance (Burfeind et al. 1996, Pollak et al. 1998, Wolk et al. 1998, Nickerson et al. 2001). In a scenario, fostering AR reactivation in a low-androgen environment (Grossmann et al. 2001), insulin resistance, and hyperinsulinemia correlates with an increased incidence of prostate cancer (Fan et al. 2007). High IGF1 levels in the serum correlate with an increased risk of prostate cancer (Pollak et al. 1998, Wolk et al. 1998), whereas IGF1 enhances AR transactivation under low/absent androgen levels (Culig et al. 1994, Orio et al. 2002) and promotes prostate tumor cell proliferation (Burfeind et al. 1996).
Endogenous AR expression as well as AR transcriptional activity is regulated by insulin via activation of the phosphatidylinositol 3-kinase (PI3K) transduction pathway (Manin et al. 1992, 2000, 2002). FOXO1, as a downstream molecule becomes phosphorylated and inactivated by PI3K/AKT kinase in response to IGF1 or insulin, and subsequently suppresses ligand-mediated AR transactivation (Fig. 1). FOXO1 is recruited by liganded AR to the AR promoters and interacts directly with the C terminus of AR in a ligand-dependent manner disrupting ligand-induced AR nuclear compartmentalization. This FOXO1 interference with AR–DNA interactions suppresses androgen-induced AR activity resulting in prostate tumor cell growth suppression (Fan et al. 2007).
An intracrine positive feedback between IGF1 and AR signaling has been implicated in prostate cancer cells. Liganded AR up-regulates IGF1 receptor expression in HepG2 and LNCaP cells, presumably resulting in higher IGF1 signaling in prostate cancer cells (Wu et al. 2007). Two AREs within the IGF1 upstream promoter activate IGF1 expression (Wu et al. 2007). In addition, androgens can control IGF signaling via modulation of IGF-binding proteins (IGFBPs) in prostate epithelial cells, while both androgens and IGF1 up-regulate IGFBP5 mRNA in androgen-responsive human fibroblasts (Yoshizawa & Ogikubo 2006). IGFBP5 initially binds IGFs with high affinity, principally by an N-terminal motif, and inhibits IGF activity by preventing IGF interaction with the type 1 receptor (Kalus et al. 1998). Taken together, this evidence supports a ‘higher-level’ interaction between AR and the IGF signaling, via recruitment of direct pathways toward activation, transcriptional regulation, and protein post-translational changes, all critical to tumor cell survival.
AR and TGFβ interactions: cell death and survival partners
Transforming growth factor-β (TGFB) is a ubiquitous cytokine that plays a critical role in numerous pathways regulating cellular and tissue homeostasis. The TGFB superfamily members regulate proliferation, growth arrest, differentiation, and apoptosis of prostatic stromal and epithelial cells, as well as the formation of osteoblastic metastases. TGFB is overexpressed in advanced prostate cancer and exerts diverse functions in stromal cells via both SMAD-dependent and SMAD-independent signaling pathways (Coffey et al. 1986, Roberts et al. 1986, Derynck & Zhang 2003, Zhu & Kyprianou 2005). Recently, cofilin and prohibitin, two novel signaling effectors of TGFB1, that serve as potential intracellular effectors of its apoptotic action were identified in human prostate cancer cells (Zhu et al. 2006). Cancer cells become refractory to the growth inhibitory activity of TGFB due to the loss or mutation of transmembrane receptors or intracellular TGFB signaling effectors during tumor initiation (Akhurst & Derynck 2001).
During prostate tumor progression to metastatic disease, TGFB1 ligand overexpression results in prooncogenic rather than growth suppressive effect. In human prostate cancer cells, TGFB signaling proceeds via ligand binding and subsequent phosphorylation of TGFBR2 receptor to the TGFBR1 kinase to SMAD activation (Zhu & Kyprianou 2005). Interaction of SMAD4, (alone or together with SMAD3), with the AR in the DNA-binding and ligand-binding domains, may result in the modulation of DHT-induced AR transactivation (Zhu et al. 2008). Interestingly, in the human prostate cancer cell lines PC3 and LNCaP, addition of SMAD3 enhances AR transactivation, while co-transfection of SMAD3 and SMAD4 actually repress AR transactivation (Kang et al. 2002). A protein–protein interaction between AR and SMAD3 has been documented both in vitro and in vivo via the transcription activation domain of AR and the MH2 of SMAD3; AR repression by SMAD3 is mediated through the MH2 domain (Hayes et al. 2001). In PC-3 prostate cancer cells, AR expression reduces the TGFB1/SMAD transcriptional activity and the growth effects of TGFB1 (in the absence of DHT), thus preventing TGFB1-induced growth inhibition and apoptosis. Furthermore, TGFB1 suppresses the E2F transcriptional activity of AR activation by DHT, an event that is associated with a reduced c-Myc expression. An ARE sequence in the TGFB promoter may provide a mechanistic basis for TGFB promoter activity toward DHT in both Huh7 and PC3/AR-expressing cells. A direct interaction between AR and TGFB1 has been causally implicated in other human tumors including hepatocarcinogenesis (Yoon et al. 2006). Androgens can inhibit TGFB1-induced transcriptional activity in prostate cancer cells (Chipuk et al. 2002), an interaction that is regulated by AR-associated protein 55 (ARA55/Hic-5; LIM protein superfamily). Overexpression of ARA55 inhibits TGFB-mediated up-regulation of SMAD transcriptional activity in rat prostate epithelial cells, as well as human prostate cells, via an interaction between ARA55 and SMAD3 mediated through the MH2 domain of SMAD3 and the C terminus of ARA55 (Wang et al. 2005).
The involvement of AR in the apoptosis outcomes of TGFB signaling in prostate cancer cells is supported by work from this laboratory. Treatment of TGFB receptor II overexpressing LNCaP TGFBR2 cells with TGFB in the presence of DHT, both cell cycle arrest and apoptosis induction are significantly enhanced over TGFB alone, through caspase-1 activation and targeting of BCL-2 (Bruckheimer & Kyprianou 2001). Enforced BCL2 expression significantly inhibits the combined TGFB and DHT apoptotic effect in prostate cancer cells (Bruckheimer & Kyprianou 2002). An androgenic contribution, with TGFB enhancement, on the epithelial-mesenchymal transition (EMT) provides an attractive mechanistic possibility in view of the assigned role of EMT during cancer metastasis (Zavadil & Bottinger 2005), with E-cadherin being considered as a potential target for such a dynamic duo.
AR and FGF interactions
The FGF family is a large family of proteins with broad spectrum of functions, including cell migration, differentiation, and angiogenesis (Ornitz & Itoh 2001). Changes in the expression of FGFs and/or their receptors are involved in prostate tumor progression toward androgen-independent disease. The estrogen receptor (ER) can regulate the synthesis of FGF2 and FGF7 in prostate cells, while stromal ER can mediate the synthesis of stromally derived growth factors, both in coordination with AR activation. AR signaling can directly dictate dramatic changes in the expression pattern of FGFs in both prostate tumor epithelial cells and stromal cells, primarily via changes in FGF1, FGF2, FGF8, and FGF10 (Saric & Shain 1998, Nakano et al. 1999, Rosini et al. 2002). Via a positive feedback, AR is up-regulated by paracrine FGF10 and synergizes with cell-autonomous activated AKT in prostate cancer cells (Memarzadeh et al. 2007). Moreover, in response to FGFs, AR facilitates FGF-induced survival of prostate cancer cells, possibly through BCL2 induction and down-regulation of AR, allowing the escape of selected clones from androgenic control (Rosini et al. 2002, Gonzalez-Herrera et al. 2006).
AR and vascular endothelial growth factor (VEGF) interactions
VEGF, originally known as vascular permeability factor, is a well-characterized angiogenic cytokine, responsible for endothelial cell proliferation, migration, and vessel assembly (Fong et al. 1995). Its value as a diagnostic tool as well as a therapeutic target for advanced metastatic prostate cancer has been examined at the molecular and translational level.
The ‘hypoxia-response’ signaling system up-regulates the expression of a network of effectors that increase the propensity of tumor cells for survival, even in this adverse environment (Anastasiadis et al. 2003). Expression of VEGF is transcriptionally induced by hypoxia-inducible factor (HIF1A) in response to oxygen changes in the microenvironment (Delongchamps et al. 2006). Androgen-stimulated growth of the glandular ventral prostate is preceded by increased VEGF synthesis, endothelial cell proliferation, vascular growth, and increased blood flow (Joseph et al. 1997, Franck-Lissbrant et al. 1998). The role of VEGF in androgen-mediated prostate vascularity was further supported by additional studies (Lissbrant et al. 2004). In prostate cancer, the effect of androgens on angiogenesis is mediated via their ability to regulate VEGF through activation of HIF1A in androgen-sensitive tumors (Boddy et al. 2005). The significant correlation between HIF1A and HIF2A expression and with AR and VEGF expression (Boddy et al. 2005, Banham et al. 2007) provides firm support for such a control system. The driving mechanism involves the direct up-regulation of VEGF-C in response to androgen depletion in prostate cancer cells (Rinaldo et al. 2007), via activation of the small GTPase, RalA; VEGF-C can increase the AR co-activator BAG-1L expression that facilitates AR transactivation. Under conditions of low-androgen levels, the intracellular reactive oxygen species induce RalA activation and VEGF-C synthesis (Rinaldo et al. 2007).
AR and growth factor interplay in the stroma
The stroma is a lead component of the prostate microenvironment contributing to tumor heterogeneity and growth dynamics. Stroma-derived fibroblasts play an active role in carcinogenesis in addition to structurally supporting the epithelial cell growth (Chung et al. 1989, 1991, Camps et al. 1990, Cunha et al. 1996). Studies in the early 1990s established that human prostate-derived stromal cells stimulate growth of prostate cancer cells in vitro and in vivo (Gleave et al. 1991). This evidence widely popularized the belief that disturbance in the epithelial–stromal interactions is most critical in the pathogenesis of prostate cancer (Hayward et al. 1998). Androgenic control during normal growth and differentiation of the prostate gland is regulated via nuclear AR in both stomal and epithelial cells (Sar et al. 1990). The close association between low-AR levels in the stroma adjacent to malignant epithelium, with a poor clinical outcome in prostate cancer patients is of high translational value (Henshall et al. 2001). Androgens increase VEGF transcription and active VEGF secretion from prostatic stroma, thus indirectly enhancing prostate cancer growth and angiogenesis (Levine et al. 1998). DHT and FGF2 can synergistically stimulate prostate stromal cell proliferation (Niu et al. 2001), while androgen depletion rapidly reduces stroma IGF1 synthesis and its action in the prostate epithelium. Close rules of compartmentalization become ‘loose’ here: although IGF1 is principally produced in the stroma and IGF-R1 in the epithelium, both are under androgenic regulation as stroma IGF1 mRNA is significantly decreased after castration, correlating with epithelial cell apoptosis (Ohlson et al. 2007).
TGFB1 is also regulator of stromal cell proliferation and differentiation, depending on the specific stromal cell type, microenvironment, and contributing activities of other growth factors (Sporn & Roberts 1992). A distinct in its complexity cross-talk between androgens and TGFB1 signaling in prostate stromal cells affects AR localization, cell proliferation, and myodifferentiation, thus defining its mechanistic contribution to the reactive stroma. AR and TGFB1 levels significantly correlate in the stromal component of prostatic intraepithelial neoplasia (Cardillo et al. 2000). Induction of rat PS-1 prostate stromal cell proliferation by androgens can be antagonized by TGFB1. Furthermore, TGFB1 triggers a cytoplasmic translocation of nuclear AR during myodifferentiation in the prostate stroma (Gerdes et al. 1998, 2004), while androgens enhance TGFB1-mediated proliferation of prostatic smooth muscle cells PSMC1 (Salm et al. 2000).
During prostate cancer progression the androgen axis engages the growth factor network to an active cross-talk toward conferring a survival and invasion advantage of prostate cancer cells. The current evidence dissecting this signaling interaction between the AR and growth factors is discussed in this review. Androgens can modify prostate cancer cell response to growth factor signals from growth inhibitory to tumor promoting during the metastatic process. A better understanding of such cross-talk between the AR axis and critical growth factor signaling in the context of the tumor microenvironment, may identify a mechanism underlying the emergence of androgen-independent prostate cancer, and provide new opportunities for therapeutic targeting of aggressive prostate tumors.
Declaration of interest
The authors hereby declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by an NIH/National Institutes of Diabetes, Digestive and Kidney Diseases R01 grant (DK-53525-08).
Acknowledgements
The authors acknowledge the assistance of Lorie Howard during the submission process.
- © 2008 Society for Endocrinology










