Somatostatin analogues in the control of neuroendocrine tumours: efficacy and mechanisms
- 1Barts and the London School of Medicine, Centre for Endocrinology, University of London, London EC1M 6BQ, UK2Institute of Endocrinology, Beilinson Hospital, Rabin Medical Center, and Sackler Faculty of Medicine, Tel Aviv 49100, Israel
- (Correspondence should be addressed to A B Grossman, Department of Endocrinology, St Bartholomew's Hospital, 5th Floor King George V Building, West Smithfield, London EC1A 7BE, UK; Email: a.b.grossman{at}qmul.ac.uk)
Abstract
Neuroendocrine tumours (NETs) represent a heterogeneous family of neoplasms, which may develop from different endocrine glands (such as the pituitary, the parathyroid or the neuroendocrine adrenal glands), endocrine islets (within the thyroid or pancreas) as well as from endocrine cells dispersed between exocrine cells throughout the digestive and respiratory tracts. The development of somatostatin analogues (SSA) as important diagnostic and treatment tools has revolutionised the clinical management of patients with NETs. However, although symptomatic relief and stabilisation of tumour growth for various periods of time are observed in many patients treated with SSA, tumour regression is rare. Possible mechanisms when this does occur include antagonism of local growth factor release and effects, probably including activation of tyrosine and serine–threonine phosphatases, and indirect effects via anti-angiogenesis. The development of new SSA, new drug combination therapies and chimaeric molecules should further improve the clinical management of these patients, as should a more complete understanding of their mode of action.
Introduction
Somatostatin (somatotrophin release-inhibiting hormone, SST) is a small polypeptide hormone present in the human body in two natural forms (14 and 28 amino acids). It is widely distributed throughout the body and binds with high affinity to five different subtypes of specific SST receptors (SSTRs) on the cell surface, which belong to the G-protein-coupled receptor family (SSTR1, SSTR2, SSTR3, SSTR4 and SSTR5; Patel et al. 1990, Lahlou et al. 2004; Table 1). SST acts as an important regulator of endocrine function by inhibiting the secretion of various hormones, such as growth hormone (GH) and all known gastrointestinal hormones (Reichlin 1987, Longnecker 1988). The very short half life of the natural compound (about 3 min in blood) has resulted in the development of synthetic analogues: short acting (octreotide, which needs to be administered subcutaneously several times per day) or long acting (octreotide long-acting release, LAR and lanreotide autogel, with a monthly administration; Heron et al. 1993).
Somatostatin receptor characteristics (chromosomal localisation of the genes encoding the five SSTR subtypes; amino acids structure; G-protein coupling and activation; effect on cAMP; signalling via tyrosine phosphatases and receptor-specific functions)
SST and its synthetic analogues may be used clinically in the treatment of a variety of neoplasms, and specifically in the therapy of neuroendocrine tumours (NETs; Grozinsky-Glasberg et al. 2008b): these comprise a family of tumours that may present with a heterogeneous range of morphological, functional and behavioural characteristics (Oberg 2005). NETs include endocrine glands (the pituitary, the parathyroid or the neuroendocrine adrenal glands), endocrine islets (within the thyroid gland or the pancreas) as well as endocrine cells distributed between exocrine cells throughout the digestive and respiratory tracts (Solcia et al. 1999, Rindi et al. 2000). The endocrine tumours of the gastrointestinal tract as well as lesions from other sites of origin were initially called carcinoids, and were classified on the basis of the anatomic site of origin into foregut carcinoids (lung, thymus, stomach, pancreas, duodenum and upper jejunum), midgut carcinoids (lower jejunum, ileum, appendix and proximal colon) and hindgut carcinoids (transverse colon, sigmoid and rectum; Vinik et al. 1989, Caplin et al. 1998, Ganim & Norton 2000). However, the latest World Health Organization (WHO) classification (Solcia et al. 2000) is based on tumour histology, tumour size and the presence or absence of local/distant metastases, and therefore divides NETs into a) well-differentiated NETs, b) well-differentiated endocrine carcinomas, c) poorly differentiated neuroendocrine carcinomas and d) mixed exocrine–endocrine carcinomas. The great majority of NETs are relatively slow growing (well differentiated), while some of them may present with an aggressive and highly malignant phenotype (poorly differentiated neuroendocrine carcinoma).
All five SSTRs bind to the natural SST, while its synthetic analogues have a limited affinity, binding mainly to SSTR2, and much less to SSTR5. The five receptors share common signalling pathways such as the inhibition of adenyl cyclase, activation of phosphotyrosine phosphatase or modulation of mitogen-activated protein kinase (MAPK) through G-protein-dependent mechanisms (Patel 1999). Due to the limited affinity of the synthetic analogues, new SST analogues (SSA) were studied and developed: pasireotide (SOM230, Novartis) is a new ‘universal’ or ‘pan-receptor’ SSA, having a high affinity for SSTR1, SSTR2, SSTR3 and SSTR5 subtypes, and is under evaluation in phase I–III trials comparing pasireotide with octreotide LAR, and exploring the utility of pasireotide in octreotide-refractory patients (Kvols et al. 2008). Its receptor-binding profile is 30–40 times higher for SSTR1 and SSTR5 than for octreotide (Bruns et al. 2002, Lamberts et al. 2002, Weckbecker et al. 2002, Shimon 2003; Table 2). Endocrine pancreatic and endocrine digestive tract tumours usually express multiple SSTR subtypes, with SSTR2 predominance generally observed. Interestingly, there is considerable variation in SSTR subtype expression between the different tumour types and even among tumours of the same type (de Herder et al. 2003).
Somatostatin receptor subtype-binding affinity of somatostatin analogues
From the various therapeutic options available for treating patients with NETs (e.g. surgery, SSA therapy, interferon-α, peptide receptor radiotherapy, chemotherapy, chemo-embolisation, etc.), few are curative and most treatments are palliative. The reason for the different biological behaviour of these tumours is unclear: why certain NETs remain localised and respond well to therapy, while others present with inoperable metastatic disease and severe hormonal symptoms, is still a matter of debate (Eriksson & Oberg 1999). The successful treatment of these diseases necessitates a multidisciplinary approach in order to control the symptoms, to stabilise or prevent further growth and, rarely, to achieve cure.
The mechanisms by which SST and its analogues exert their effects on the NET cells are complex but poorly understood. SST is known to be able to inhibit different cellular functions, such as secretion, motility and proliferation. SST exerts its activity by binding to cell- and tissue-specific receptors, but its action depends on the site of formation: within the central nervous system, SST acts as a neurotransmitter, while its hypothalamic–hypophyseal transfer qualifies it as a neurohormone. In other tissues, SST has a paracrine (regulating adjacent cells) or an autocrine (self-regulation) activity (Reichlin 1987, Schally 1988, Besedovsky & del Rey 1996, Zaki et al. 1996, Low 2004). Finally, after secretion into the intestinal lumen, it can behave as a ‘lumone’, acting directly on the gut (Krulich et al. 1968, Rivier et al. 1982). The SSTRs are expressed in about 80–90% of NETs; their expression is the basis for the use of SSAs in the treatment of these tumours (Reubi et al. 1990).
The SSAs have been used in the treatment of NETs, and specifically of gastroenteropancreatic NETs (GEP-NETs), for many years: they may help in alleviating symptoms associated with functional tumours (e.g. carcinoid syndrome, Verner–Morrison syndrome) or in inhibiting tumour progression in patients with advanced disease. The anti-tumour effect of SSAs may include both a cytostatic (growth arrest) and a cytotoxic (pro-apoptosis) mechanism (Balaban & Severs 1992, Bousquet et al. 2001, Wulbrand et al. 2002, Ferrante et al. 2006, Pyronnet et al. 2008). However, there is still little known regarding the anti-proliferative role of SSA in NETs, although increasing data suggest that such analogues can be tumouristatic, at least in some circumstances (Jensen 2000).
By contrast, in pituitary tumours-secreting GH, SSAs have been shown to be more effective in causing tumour regression (Zatelli et al. 2006), although there may be dissociation between the anti-secretory and growth-inhibitory effects. This dissociated effect of SSA was demonstrated in one study using primary cell cultures from an octreotide-resistant acromegalic patient (Resmini et al. 2007): the significant anti-proliferative effect of octreotide was shown to be related to the higher expression of SSTR5, while the negligible anti-hormonal effect was directly related to the lower expression of SSTR2 by tumour cells.
In this review, we summarise the literature regarding the mechanism of action of SST and its analogues, specifically their anti-proliferative and/or anti-secretory effects, which are not always concordant. The data are presented in two separate parts: first, in which preclinical data on the SSA anti-proliferative, apoptotic and anti-angiogenetic effects are summarised (using reports on tissues other than NETs as well, as data specific to NETs are few) and second, in which the clinical symptomatic, biochemical and tumour-shrinkage effects of SST and its analogues are reviewed.
Overview of the SST anti-tumour mechanisms – preclinical data on anti-proliferation, apoptosis and anti-angiogenesis
The effect of SST on cell cycle progress and growth-promoting factors
SST-induced cell growth arrest is poorly understood. SST and its analogues were demonstrated to have direct (i.e. SST acts on the tumour cell itself and not via other tissues such as vessels) anti-proliferative effects in a variety of tumour cells by inhibiting the mitogenic signalling of growth factor receptor kinases, but also by inducing apoptosis (Thompson 1999, Liu et al. 2000, Lahlou et al. 2004). They also may inhibit the secretion of insulin-like growth factor-I, which has been thought to be involved in recurrence, growth and aggressiveness of some endocrine and non-endocrine tumours (Furukawa et al. 2005).
The SSTR-mediated effect on tumour cell proliferation has been considered to include several mechanisms, related to both specific receptor and cell subtype. Different SSTRs (SSTR1, SSTR2, SSTR4 and SSTR5) have been implicated in vitro in the G1–G0 cell cycle blockade, the apoptotic effect of SST being mediated through SSTR2 and SSTR3. The effect on cell proliferation may be sometimes opposite, depending on the receptor splice variants (SSTR2A or SSTR2B) that are activated (Alderton et al. 1998). As a consequence of retinoblastoma gene product (Rb) hypophosphorylation and G1-phase cell cycle arrest, ligand-activated SSTR1, SSTR2A, SSTR4 and SSTR5 may suppress the mitogenic signal of serum growth factors (Sharma et al. 1999). SSTR2 upregulation of a cyclin-dependent kinase inhibitor p27 (Kip1) induces cell cycle arrest; the cytoplasmic protein tyrosine phosphatase, Src homology 2-containing protein SHP-1, is required for maintaining high inhibitory levels of p27 (Kip1), being a critical target of the insulin and SST signalling cascade (Pages et al. 1999).
Preclinical data regarding SSA anti-proliferative effects in other tumours and cell lines
-
Initially, the effects of SST and of its analogue octreotide on proliferation of GH3 pituitary tumour cells were investigated in vitro showing a significant, but transient, inhibition on GH3 cell growth (Pelicci et al. 1990). In GH3 rat pituitary tumour cells, SSAs induced G0–G1 cell cycle arrest, preventing DNA synthesis (Cheung & Boyages 1995).
In pituitary cells (human samples as well as in GH3 cell line), the upregulation of p27 protein levels and the inhibition of phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2) were shown, suggesting that SST-mediated growth inhibition is associated with the downregulation of pERK and the upregulation of p27 (Hubina et al. 2006).
-
In MCF7 human mammary tumour cells, transient G2/M blockade and apoptosis were demonstrated (Pagliacci et al. 1991, Sharma et al. 1996). In these cells, octreotide had cytotoxic effects leading to apoptosis, with a rapid time-dependent induction of wild-type p53 and an increase in B-cell lymphoma 2 (BCL2)-associated X protein; there was no G1 cell cycle arrest in these cells during the administration of octreotide, as suggested by the decrease in G1/S ratio and the lack of induction of tumour suppressor retinoblastoma protein, pRb, or of the regulator of cell cycle progression at G1, p21. These data support the idea of using SSA in the treatment of SSTR-positive breast cancers expressing wild-type p53 (Sharma & Srikant 1998).
-
In pancreatic cancer cells, the absence of expression or coupling of the receptors involved in the anti-proliferative process may explain the dissociation observed sometimes between the anti-secretory and the anti-tumour effects of different SSAs; coupling to membrane tyrosine phosphatases (SHP-1 and SHP-2) is the main transduction pathway that has been implicated in the SSTR-mediated anti-proliferative effects (Buscail et al. 2002). In human pancreatic adenocarcinoma, it was shown that the cells lose the ability to express SSTR2. Reintroducing this receptor into the pancreatic cancer cells by stable expression leads to a constitutive activation of the SSTR2 gene and evokes a negative feedback loop, inhibiting cell proliferation. This may suggest that SSTR2 gene transfer might be considered as a possible novel therapy for pancreatic cancer (Rochaix et al. 1999).
-
In Chinese hamster ovary cells, which express SSTR1, it has been shown that SST can stimulate the tyrosine phosphatase SHP-2, activate the MAPK cascade and induce the p21 cyclin-dependent kinase inhibitor; as a result, SST induces cell growth arrest in these cells (Florio et al. 1999).
Preclinical data regarding SSA anti-proliferative effects in NETs
Regarding neuroendocrine tissues, the data are scarce: in the human medullary thyroid carcinoma (MTC) TT cell line, which express all five SSTRs, it was demonstrated that SST and its analogues inhibit cell proliferation via SSTR2 (Zatelli et al. 2001, 2002); it was shown that this inhibitory effect on cell proliferation is partially produced by SSTR2-mediated stimulation of the tyrosine phosphatase SHP-1 (Zatelli et al. 2005). The same authors demonstrated that SST-induced SHP-1 activation downregulates MAPK signalling in TT cells, decreasing cell proliferation.
Using the same MTC TT cell line, Tagliati et al. (2006) assessed the effects of SST and its selective SSTR2 agonist BIM-23120 on cell cycle protein expression, paying special attention to cyclin D1 and its associated kinases. It was shown that both drugs were able to reduce cell proliferation and DNA synthesis as well as to induce a delay in the cell cycle in G2/M phase; cyclin D1 levels decreased, with a parallel increase in phospho-cyclin D1 levels, suggesting protein degradation. These data suggest that a decrease in cyclin D1 levels may be an important element in the SST SSTR2-mediated anti-proliferative effect on TT cell proliferation.
SST's effect on tumour angiogenesis
Angiogenesis is a fundamental process in the context of tumour growth, and one of the main factors involved in the appearance of new tumour vessels is vascular endothelial growth factor (VEGF). SST and its analogues may inhibit the production and secretion of many angiogenic factors, thereby reducing tumour growth rate (Barrie et al. 1993). It has been demonstrated that octreotide-induced inhibition of angiogenesis is G-protein, calcium- and cAMP dependent, and is protein kinase C (PKC) and tyrosine phosphatase independent (Patel et al. 1994). SSTR expression has been demonstrated in peritumoral vessels in different tumour types, and it appears to be unrelated to the receptor expression in the tumour cells (Reubi et al. 1997). There are several studies that support the potential anti-angiogenic effect of SST and its analogues. One study hypothesised that non-proliferating human vascular endothelial cells do not express SSTR2 but that this receptor is expressed when the endothelial cells begin to grow (Watson et al. 2001); the SSTR2 gene was expressed, and the presence of SSTR2 on proliferating angiogenic vessels was confirmed, by immunohistochemical staining and in vivo scintigraphy, suggesting that SSTR2 may be a specific target for anti-angiogenic therapy with SSTR2-binding SSAs conjugated to radioisotopes or cytotoxic agents.
SST was reported to inhibit Kaposi sarcoma (KS) cell (KS-Imm) xenografts through an anti-angiogenic activity; it was shown that SST blocks the growth of established KS tumours with the same efficacy as the cytotoxic drug adriamycin. Whereas KS-Imm cells do not express SSTRs, endothelial cells express several SSTRs, particularly SSTR3. It was shown that endothelial nitric oxide synthase (eNOS) inhibition was an important prerequisite for the anti-angiogenic effects of SST, and that SST is a powerful anti-tumour angiogenesis agent through SSTR3-mediated inhibition of both eNOS and MAPK activities (Florio et al. 2003).
The effects of SST and its pan receptor ligand pasireotide (SOM230) on VEGF secretion, cell viability and proliferation were assessed in a study using human non-functional pituitary adenoma (NFPA) primary cultures (Zatelli et al. 2007): 25 adenomas were examined by RT-PCR for the expression of SSTRs, VEGF and VEGF receptors 1 and 2. VEGF secretion and cell viability were reduced, and both drugs completely abrogated the promoting effects of VEGF on cell viability. These data demonstrate that pasireotide can inhibit NFPA cell viability by inhibiting VEGF secretion, and suggest that the pasireotide could be used in the therapy of selected NFPAs.
Therefore, SSAs may suppress tumour growth either directly, through their effect on SSTR expressing cells, or indirectly, via inhibition of angiogenic factors such as VEGF (Albini et al. 1999, Mentlein et al. 2001).
SSAs and tumour cell proliferation pathways
The PI3K/Akt/mTOR/p70S6K pathway
One of the multiple possible pathways involved in tumour cell proliferation, which has been increasingly studied during the last few years, is the phosphatidylinositol-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR)/p70S6K pathway.
In many cancers, overexpression and activation of PI3K, which leads in turn to activation of Akt, has been demonstrated: both kinases promote cell growth and proliferation, survival and increased motility, and promote increased cell size and response to nutrient availability, tissue invasion and angiogenesis (Altomare & Testa 2005). It has been shown that they induce tumour progression in breast, ovarian, prostate, pancreatic and thyroid cancers (Cheng et al. 1996, Vasko et al. 2004, Bellacosa et al. 2005, Wu & Huang 2007). The activation of Akt stimulates downstream proteins including mTOR and p70S6K (the serine–threonine kinase of p70S6), which both play a significant role in cell growth and proliferation. Tuberous sclerosis complex 2, tuberin (TSC2), is an important intermediate in this signalling cascade. TSC2 phosphorylation by activated Akt will induce dissociation of the TSC1–TSC2 complex, inactivating the constitutive inhibition of Ras homologue enriched in brain (Rheb) and thereby releasing the inhibitory effects of the TSC1–TSC2 complex on mTOR (McManus & Alessi 2002). The molecules required for positive regulation of Rheb have not been identified as yet. One recent study showed that a conserved protein, translationally controlled tumour protein (TCTP), is an essential novel component of the TSC-Rheb pathway and a direct regulator of Rheb; human TCTP shows similar biochemical properties compared with Drosophila TCTP (dTCTP), and can rescue dTCTP mutant phenotypes, suggesting that the function of TCTP in the TSC pathway is evolutionarily conserved (Hsu et al. 2007). The serine–threonine kinase mTOR has emerged as a major effector of cell growth and proliferation via the regulation of protein synthesis and inhibition of apoptosis (Cheng et al. 2004, Hay & Sonenberg 2004, Petroulakis et al. 2006). Recently, it was shown that mTOR is also necessary for the maintenance of mitochondrial oxidative function (Cunningham et al. 2007). This important signalling pathway has been little studied in NETs.
In a study on pituitary tumour cells, treatment with the SSA octreotide decreased the tyrosine phosphorylation levels of the PI3K regulatory subunit p85, induced dephosphorylation of phosphoinositide-dependent kinase 1 (PDK1) and Akt, and activated glycogen synthase kinase-3β. In this case, SSAs were thought to produce their anti-proliferative action by acting on the PI3K/Akt signalling pathway and increasing Zac1 gene expression (Theodoropoulou et al. 2006).
In a recent study (Grozinsky-Glasberg et al. 2008a), we treated INS1 cells, a rodent-derived insulinoma cell line, with the SSA octreotide, with RAD001 (everolimus, a derivative of the mTOR inhibitor rapamycin) and with their combination at different times and using a variety of concentrations; we looked for their effect on cell proliferation and on different phosphorylation sites in the Akt/mTOR/p70S6K pathway, specifically to explore the mode of action of octreotide in causing tumoristasis. Our results showed that octreotide and RAD001 have significant anti-proliferative effects. Octreotide inhibited the phosphorylation of TSC2, mTOR and p70S6K, while the phosphorylation of Akt was unaffected (Fig. 1). While RAD001 is known to interact with the raptor site of mTOR, we suggested in this study that RAD001 may interact with the same pathway at a site or sites similar to octreotide, possibly the principal site involved being the inhibition of phosphorylation of TSC2, thereby preserving the TSC2 suppressive effect on mTOR (Fig. 2). In most cell types, Akt is a point of convergence for important signalling pathways, which may include PI3K, which recruits Akt to the cell membrane and induces its Thr308 site phosphorylation by activating the PDK1, or the MAPK-activated protein kinase 2 and the phorbol-12-myristate-13-acetate activation of PKC, which all may be involved in the phosphorylation of the Akt Ser473 site (Luo et al. 2003, Woodgett 2005, Barragan et al. 2006, Theodoropoulou et al. 2006). These effects of octreotide all seem to imply an activation of a serine–threonine phosphatase, but precise evidence in favour of this speculation is not available.
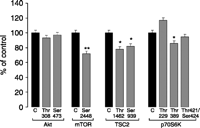
Effect of octreotide on phosphorylation of different residues on Akt/TSC2/mTOR/p70S6K pathway. In insulinoma cell line (INS1), octreotide has a non-significant effect on Akt phosphorylation sites (Thr308 and Ser473). However, octreotide significantly suppressed mTOR phosphorylation at Ser2448 site after 30-min treatment (P=0.015), and significantly decreased TSC2 phosphorylation at both Ser939 (P=0.0133) and Thr1462 (P=0.0124) phosphorylation sites. Moreover, p70S6K phosphorylation at Thr389 site was significantly inhibited by octreotide (P=0.048), while no effect was observed on the other two sites (Thr229 and Thr421/Ser424). Values are shown as average and s.e.m. of three replicates values from the same experiment, which were confirmed by three different experiments S Grozinsky-Glasberg & G Franchi, unpublished data. *P<0.01 vs. control.
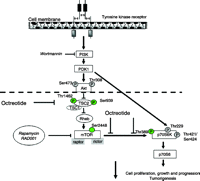
The Akt/TSC2/mTOR/p70S6K pathway and the proposed sites of action for octreotide. Integration of nutrient and growth factors regulates mTOR-dependent downstream signalling. PI3K localizes Akt to the membrane where it can be phosphorylated and activated by PDK1. Akt is activated by phosphorylation at Thr308 or at Ser473 sites. Activated Akt phosphorylates TSC2 (Thr1462 or Ser939), resulting in TSC1/TSC2 complex instability and inhibition of the tumour suppressor function of the TSC2. Rheb, a small tyrosine phosphatase, is inhibited by the TSC2/TSC1 complex and positively modulates mTOR function. Phosphorylation of mTOR at the Ser2448 site promotes the phosphorylation of p70S6K (Thr389 site), resulting in the activation of p70S6K. p70S6K may also be activated by phosphorylation at the Thr229 catalytic site by a PI3K/PDK1-dependent, mTOR-independent, mechanism. The third phosphorylation site on p70S6K depicted in the figure (Thr421/Ser424) is an autophosphorylation site localised in the autoinhibitory region of the kinase; its phosphorylation stimulates p70S6K activity. Increased p70S6K action promotes cell growth and cell cycle progression. Octreotide inhibited the phosphorylation of TSC2, mTOR and p70S6K, while the phosphorylation of Akt was unaffected (Grozinsky-Glasberg et al. 2008a). Arrows depict activation, while bars depict inhibition. The phosphorylation sites of TSC2 depicted in italic bold represent inhibitory sites; all others sites are stimulatory.
The ERK-MAPK pathway and possible interrelation with the Akt/mTOR pathway
While one of the most defined cell survival signalling cascade in many cell types involves PI3K and its downstream target Akt (protein kinase B), Akt is certainly not the only PI3K-activated molecule involved in cell survival (Ballif & Blenis 2001). Among others, PI3K can activate survival kinases such as the PKC and consequently the ERKs family members (Wooten 1999). Activation of cytosolic receptors with growth factors, cytokines, hormones or the proto-oncoprotein Ras, results in activation of the MAPKs and the ERK-regulated kinases. Signalling by these protein kinases alters the activity of several proteins involved in cell adhesion and motility, cell proliferation, differentiation and survival (Fig. 3). However, the molecular ways from MEK (‘MAPK/ERK kinase’) to promote cell survival in the NETs, the interrelation with the Akt/mTOR pathway, and the role, if any, of SSAs on the activity of these kinases, remain to be elucidated.
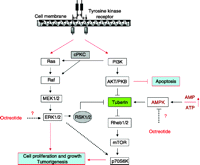
The Ras-Raf-MEK-ERK (mitogen activated protein kinase, MAPK) and PI3K-AKT-mTOR signalling pathways represent significant and promising molecular targets for effective treatment of neuroendocrine tumours. Tyrosine kinase receptor has at least two survival signals that are able to induce cell proliferation and to protect cancer cells from apoptosis, namely PI3K/AKT and MAPK/ERK signalling pathways. Signalling by these protein kinases alters the activity of several proteins that regulate cell adhesion and motility, differentiation and proliferation, and cell survival. While tuberin (TSC2) seems to be a critical point of convergence for these two pathways, important details regarding its regulation by somatostatin analogues remain to be solved, such as the effects of these drugs on ERK or AMPK activity. PI3K, phosphoinositide-3-kinase; AKT/PKB, protein kinase B; Rheb, Ras homologue enriched in brain; mTOR, mammalian target of rapamycin; 70S6K, 70 kDa S6 protein kinase; MEK, MAPK/ERK kinase; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinases; PKC, protein kinase C; RSK, p90 ribosomal S6 kinase; AMPK, AMP-activated protein kinase and ?, possible sites of action for octreotide.
In summary, the mechanisms which appear to be involved in SST's inhibition of tumour proliferation are complex; consideration has to be given to the idea of achieving an increased anti-tumour activity by using SSAs that bind to more than one receptor subtype (e.g. to SSTR2 and SSTR5, or to SSTR1–SSTR2–SSTR3–SSTR5, as shown with the pan receptor agonist SOM230/pasireotide; Bruns et al. 2002, Jaquet et al. 2005, Kvols et al. 2005). Chimaeric molecules, such as the SSTR2 and dopamine receptor D2R molecule (dopastatin; Saveanu et al. 2002), or conjugated peptides, as for example, the camptothecin–SST conjugate peptide (Moody et al. 2005), have also shown some promising in vitro tumour inhibitory effects.
The symptomatic, biochemical and tumour-shrinkage effects of SST and its analogues – clinical data
Symptomatic and biochemical effects of SSAs on different NETs
GEP-NETs constitute ∼2% of all malignant tumours of the gastrointestinal system (Moertel 1987). Midgut carcinoids, which originate from serotonin-producing enterochromaffin cells, constitute the largest group, while the second largest group includes endocrine pancreatic tumours. Pancreatic tumours may be subdivided depending on the predominant hormone production and the clinical picture. In order to diagnose these tumours, the index of suspicion must be high, as patients may have symptoms for many years before the diagnosis is made.
The clinical use of the SSA octreotide in the treatment of NETs has been reported since the 1980s, and it was based on both cytotoxic and cytostatic effects: it was shown to influence cell growth and induce apoptosis, particularly at high doses (Garcia et al. 2002).
-
a. Endocrine tumours of the gastric mucosa (gastric carcinoids), which originate from enterochromaffin like (ECL) cells, may be divided into four distinct categories: type 1, the majority of gastric carcinoids (∼75%), are associated with chronic atrophic gastritis; type 2 (between 5% and 10%) are associated with the Zollinger–Ellison syndrome and occur almost exclusively in the context of multiple endocrine neoplasia type 1; type 3 gastric carcinoids (15–25%) are sporadic and highly aggressive and type 4 are poorly differentiated endocrine carcinomas (Gough et al. 1994, Soga 1997, Kulke & Mayer 1999, Rindi et al. 1999). A fifth group can be added to this classification, the ghrelin-secreting gastric carcinoid tumours (Tsolakis et al. 2004).
Over the last few years, SSAs have been increasingly used in the treatment of patients with type 1 or 2 gastric carcinoids. While treatment with proton pump inhibitors is very effective in reducing hypergastrinaemia-induced gastric acid hypersecretion (Tomassetti et al. 2005), it does not improve the ECL cell hyperplasia. In a case report of a patient with multiple type 1 gastric carcinoids, treatment with the long-acting SSA octreotide LAR for a period of 9 months induced normalisation of serum gastrin levels and permanent disappearance of the tumours (Prommegger et al. 2003). In another study (Fykse et al. 2004), five patients with hypergastrinaemia and gastric carcinoids were treated for a period of 1 year with monthly injections of octreotide LAR; at the end of the study, although gastrin levels did not totally normalise, there was a significant reduction in tumour load, ECL cell density and normalisation of circulating chromogranin A levels, indicating a possible direct anti-proliferative effect of the treatment. Furthermore, another study presented three patients suffering from Zollinger–Ellison syndrome, who were treated with lanreotide or octreotide for a period of 1 year, showing a significant reduction in the gastrin levels and no evidence of the tumour at the end of the study (Tomassetti et al. 2000a). Although the number of patients included in these studies is small, these results suggest that the SSAs have an important inhibitory effect on gastrin secretion as well as on the formation of these tumours. However, in patients with gastric carcinoids type 3 or poorly differentiated endocrine carcinomas, SSA treatment may be considered only as palliative, reducing symptoms related to the carcinoid syndrome.
-
b. Midgut carcinoids are the most common of the GEP-NETs (∼50% of all carcinoid tumours), affecting about five to seven new patients per million population per year. They are usually slow growing and the clinical presentation may be related to pain, due to the tumour mass effect or to fibrosis in the mesentery. The ‘carcinoid syndrome’ (defined as watery diarrhoea, flushing, right-sided heart failure and bronchial constriction) is reported in about 10–15% of these patients and is considered to be produced by the tumour hypersecretion of a variety of endocrine substances, the most frequent of which are serotonin (5-hydroxytryptamine) and the tachykinins (Kulke & Mayer 1999). Surgery is rarely curative, as the majority of these patients have metastatic disease at the time of diagnosis; therefore, medical treatment has to be considered (Oberg 2002).
Administration of SSAs, at variable dosages (from 100 μg twice a day to 200 μg thrice a day for octreotide, 10–30 mg octreotide LAR every 4 weeks or 30 mg depot lanreotide every 10–14 days), may significantly improve symptoms related to the carcinoid syndrome, such as diarrhoea or flushing (between 38 and 88% in different studies); significantly lower levels of urinary hydroxyindoleacetic acid (5-HIAA), the metabolite of serotonin, were also observed in the treated patients (Kvols et al. 1986, Vinik et al. 1986, Oberg et al. 1991, Wymenga et al. 1999, Rubin et al. 1999, O'Toole et al. 2000). In a multicentre study of 33 patients diagnosed with the carcinoid syndrome, treatment with lanreotide (30 mg i.m. every 10 days) was compared with octreotide administration (200 μg s.c. twice or thrice daily) in terms of patient preference and efficacy in controlling symptoms (O'Toole et al. 2000). Disappearance or improvement symptoms occurred in 53.8% of the patients treated with lanreotide, while they were observed in up to 68% of those on octreotide. No significant differences were found in terms of quality of life, and both drugs were equally effective in reducing urinary 5-HIAA levels and plasma serotonin levels. In another study of 71 patients with the carcinoid syndrome, six treatments of prolonged release lanreotide autogel were administered in various doses (60, 90 or 120 mg) depending on symptom response over a period of 6 months (Ruszniewski et al. 2004); 65% of the patients with flushing and 18% of diarrhoea patients achieved a more than 50% reduction of symptoms from baseline, with significant reductions in urinary 5-HIAA and blood chromogranin A levels. A randomised double-blind trial, in which octreotide LAR at 10, 20 and 30 mg every 4 weeks was compared with open-label s.c. octreotide every 8 h for the treatment of carcinoid syndrome, showed that the efficacy of both treatment arms was the same once plasma octreotide steady-state concentrations were achieved (Rubin et al. 1999). This study also suggested that the starting dose for octreotide LAR should be 20 mg.
-
c. Endocrine pancreatic tumours may be classified according to their secretory ability into functioning or non-functioning tumours. The non-functioning tumours constitute the largest group, representing ∼50% of the endocrine pancreatic tumours (Evans et al. 1993); following in incidence are the insulinomas (25%; de Herder et al. 2006) and gastrinomas (15%; Roy et al. 2000), while the remaining 10% include vasoactive intestinal polypeptide (VIP)-omas, glucagonomas and somatostatinomas (Soga & Yakuwa 1998, 1999). Most of these tumours are sporadic, while about 15–30% are hereditary and appear in the context of multiple endocrine neoplasia type 1 (MEN1) or von Hippel–Lindau syndromes.
Insulinoma is a rare but important cause of endogenous hypoglycaemia, occurring with an incidence of about 1/million population per year (Service et al. 1991). Treatment with SSAs can improve (inhibition of insulin release) or worsen (profound suppression of counter-regulatory hormones GH and glucagon) the hypoglycaemia-associated symptoms (Maton 1993). Recently, the efficacy of octreotide on hypoglycaemia was assessed in a study of 17 patients with insulinoma (Vezzosi et al. 2005): in more than 50% of the patients, the drug was effective in hypoglycaemia control. A positive s.c. short octreotide test (100 μg octreotide s.c. in fasting patients, with improvement in hypoglycaemia) was a better marker of the therapeutic response compared with a pretreatment positive scintigraphy.
Regarding gastrinomas, proton pump inhibitors are currently the therapy of choice for the control of gastric acid-associated symptoms, which dominate the clinical picture (Metz et al. 1993), but SSAs may also occasionally be used. Glucagonomas are rare slow-growing tumours, originating in the α-cells of the pancreas. Most of them are sporadic, and rarely they may be associated with familial syndromes, such as MEN1 or familial adenomatous polyposis (Chastain 2001). SSAs may be useful for alleviating symptoms related to the characteristic skin rash (necrolytic migratory erythema) or diarrhoea (Wermers et al. 1996, Casadei et al. 1999, Tomassetti et al. 2000b).
Somatostatinomas are very rare tumours, originating either in the pancreas or in the small intestine (Soga et al. 1990, Soga & Yakuwa 1999); the symptoms are usually related to SST hypersecretion (hyperglycaemia, cholelithiasis, diarrhoea and steatorrhoea, hypochlorhydria) or to the mass effect (Soga & Yakuwa 1999). Treating patients with symptoms related to elevated SST levels with a SSA is a paradoxical concept. However, in a study of three patients with metastatic somatostatinomas, octreotide treatment was effective in reducing plasma levels of SST and in improving related symptoms (Angeletti et al. 1998).
VIP-omas are rare VIP-secreting pancreatic tumours. VIP hypersecretion will induce hypersecretion of water and electrolytes by the intestinal mucosa, producing a pathognomonic clinical syndrome (Verner–Morrison syndrome, characterised by watery diarrhoea, hypokalaemia, achlorhydria and metabolic acidosis; Bloom et al. 1973, Schwartz et al. 1974). The treatment includes i.v. fluid and electrolyte replacement, while octreotide administration will essentially control symptoms in more than 90% of patients (O'Dorisio et al. 1989). In a recent retrospective review, octreotide was very successful as an adjuvant therapy for symptoms control and for reducing the serum-elevated VIP levels in four cases of VIP-oma (Ghaferi et al. 2008), improving the diarrhoea and the electrolyte imbalance. In treatment failures, high-dose corticosteroids may be used.
In a phase II open label, multicentre study including 21 metastatic carcinoid tumours patients whose symptoms (diarrhoea and flushing) were refractory to octreotide LAR, pasireotide at dosages between 450 and 1200 μg twice a day effectively controlled symptoms in one-third of these patients (Kvols et al. 2005). This suggests that at least some of the refractoriness of carcinoid tumours to octreotide may be due to the expression of SSTRs other than SSTR2, and thus that pasireotide (especially in long-acting formulations currently under development) may be useful in such patients.
MTC, which originates in the parafollicular C cells, is a rare tumour of the thyroid (3–10% of all thyroid carcinomas). MTC may synthesise and secrete calcitonin in high amounts as well as other peptides such as carcinoembryonic antigen (CEA), neuron-specific enolase, chromogranin A or adrenocorticotrophin (ACTH) resulting in diarrhoea, facial flushing or Cushing's syndrome (Raue 1998, Kebebew et al. 2000). The initial and the only potentially curative treatment of choice is total thyroidectomy with central lymph node dissection (Giuffrida & Gharib 1998). The data regarding the effect of SSAs in the treatment of symptomatic MTC are controversial: in one study, this treatment significantly improved symptoms such as diarrhoea, weight loss or malaise in all of the three patients with metastatic MTC, with a parallel decrease in the calcitonin and CEA levels (Mahler et al. 1990). In another study, 14 post-thyroidectomy metastatic MTC patients were treated with continuous s.c. infusion of 500 μg/day octreotide, for 90 days (Modigliani et al. 1992): continuous infusion of octreotide did not induce any morphological improvement or a significant decrease in calcitonin levels; in four patients, calcitonin levels fell during treatment (between 15% and 50%), while in nine patients, calcitonin increased (from 22% to 130%) after cessation of therapy. In patients with advanced metastatic disease, the administration of octreotide combined with interferon was studied: in a study of eight patients with advanced MTC, patients received octreotide (at a starting dose of 150 μg/day s.c. for 6 months, followed by a dose of 300 μg/day s.c. for another 6 months) combined with recombinant interferon-α-2b (rIFN-α-2b) (at a dose of 5 million IU/day i.m. thrice a week, for 12 months; Lupoli et al. 1996). This combination induced a significant improvement in the symptoms of some of the treated patients (diarrhoea, in four patients, and flushing, in one). The calcitonin levels decreased maximally after 3 months in four patients, and the CEA levels decreased in all patients during treatment.
Bronchial carcinoid tumours belong to the foregut carcinoids, accounting for about 2.5% of all pulmonary neoplasms and for 12–15% of carcinoid tumours overall. They originate from the neuroendocrine cells of bronchial mucosa, and may present with a wide range of clinical and biological behaviours, including their potential to synthesise and secrete peptide hormones (such as ACTH, serotonin, SST or bradykinin). The latest WHO morphological classification of lung NETs include low-grade typical carcinoid, intermediate-grade atypical carcinoid, the high-grade small-cell lung carcinoma (SCLC) and large-cell neuroendocrine carcinoma (Beasley et al. 2005). While many of the patients with bronchial carcinoids (13–51%) are incidentally detected on routine chest X-ray, presenting symptoms may include cough, haemoptysis, dyspnoea, wheezing, chest pain and recurrent pulmonary infections; the carcinoid syndrome (with flushing, diarrhoea, wheezing and elevated urinary 5-HIAA) occurs in 2–12% of the patients, mostly in those displaying liver metastases (Dusmet & McKneally 1994, Soga et al. 1999). An atypical histamine-induced carcinoid syndrome (with severe generalised flushing, swelling, lacrimation, asthma and diarrhoea) may be sometimes observed, while Cushing's syndrome, due to ectopic secretion of ACTH or corticotrophin-releasing hormone, is seen in 2–6% of bronchial carcinoid patients (Soga et al. 1999).
The effect of octreotide in treating bronchial carcinoids is mainly symptomatic (Granberg et al. 2001). The combination of α-interferon and octreotide may produce efficient symptomatic relief, and may be tried in patients unresponsive to octreotide alone. However, s.c. administration of octreotide at a daily dosage of 1500 μg controlled the carcinoid syndrome-associated symptoms in all of the seven patients included in another study (Filosso et al. 2002). Moreover, patients with Cushing's syndrome due to ectopic ACTH secretion from lung carcinoids may be treated with octreotide, which has been shown to be effective in reducing the circulating ACTH levels in some cases (Hearn et al. 1988).
The thymus is one of the rarest sites for the development of NETs. Thymic carcinoid tumours usually carry a poor prognosis, being frequently metastatic, and are commonly associated with ectopic ACTH production but not with the carcinoid syndrome (Moran & Suster 2000). While eight cases of thymic carcinoids were first described in 1972 as a different entity from thymic carcinomas (Rosai & Higa 1972), about 150 cases have been reported since. These tumours may rarely appear in the context of MEN1 (Rosai et al. 1972). There are a number of case reports in which SSAs were of value in the detection and symptomatic relief in patients with thymic carcinoid tumours associated with ectopic GH-releasing hormone (Boix et al. 2002) or ectopic ACTH secretion (Matejka et al. 1996).
Ovarian carcinoids are very rare (0.52–1.7% of ovarian tumours in different series), presenting with pain in the pelvic area or during defaecation (Davis et al. 1996). Some of them are cystic teratomas, having a more benign course (a 5-year survival of almost 100%), while others have a 5-year survival rate of about 84%. The carcinoid syndrome is present in ∼30% of patients, and there are also reports regarding ovarian carcinoids ectopically secreting ACTH and inducing the clinical picture of Cushing's syndrome (Schlaghecke et al. 1989). The treatment of choice for these tumours is surgical excision; therefore, data regarding the use of octreotide are limited mostly to carcinoid syndrome-associated symptom improvement, or to the intraoperative use in patients with carcinoid associated right-sided heart failure (Watson et al. 1990, Vergani et al. 1998).
Phaeochromocytomas and paragangliomas arise from chromaffin cells and may occur in sporadic or familial forms (associated with MEN2A or 2B, von Hippel–Lindau syndrome, neurofibromatosis 1, Carney's triad or mutations of succinic dehydrogenase subunits C, D and particularly B; Goldstein et al. 1999, Astuti et al. 2003, 2004, Mhatre et al. 2004, Neumayer et al. 2007). Phaeochromocytomas originate in the adrenal medulla (Shapiro & Fig 1989), while paragangliomas derived from the paraganglia (either sympathetic, localised mainly in the retroperitoneum and thorax, or parasympathetic, occurring in the area of the aortic arch, neck and skull base). While the catecholamines (adrenaline, noradrenaline and dopamine) are the main secretory products of chromaffin cells, a number of other hormones have been described in association with functioning catecholamine-secreting tumours: secretion of ACTH, inducing Cushing's syndrome; substance P, tachykinins and histamine-inducing hypotension; VIP and calcitonin gene-related peptide that may produce flushing (Bravo & Tagle 2003).
Surgical excision of the tumour, performed after symptom stabilisation using specific anti-hypertensive treatment, is the treatment of choice in order to obtain disease-free long-term survival (Bravo 2002). Short- or long-term administration of SSAs has not been shown to be of any clear benefit, although rarely biochemical responses have been observed (Kopf et al. 1997), being capable of lowering the levels of noradrenaline, but with no consistent effect on blood pressure (De Invitti et al. 1993, Lamarre-Cliche et al. 2002). However, another study demonstrated that sometimes octreotide may control the blood pressure before surgery in some patients where phaeochromocytoma-induced hypertension is uncontrolled (Koriyama et al. 2000).
Clinical data on SSAs and tumour shrinkage in NETs
There is still little known about the anti-proliferative effect of SSAs on the growth of the midgut carcinoids; partial/complete responses have been described in fewer than 10% of the patients, while stabilisation of tumour growth was observed in 24–57% of the patients (Plockinger et al. 2004). Moreover, it was suggested that the anti-proliferative effect of these drugs may be dose related. The administration of SSAs in regular doses induced tumour growth stabilisation of in about 40–50% of the patients in different studies (Saltz et al. 1993, Arnold et al. 1996, Di Bartolomeo et al. 1996), and this effect persisted for varying periods of time (between 2 and 60 months). Tumour regression was partial in 2 out of 38 patients included in one of these studies (Di Bartolomeo et al. 1996), but no tumour regression was described in the other two studies.
There are studies that suggest that using a higher than usual dose of SSAs may be more effective in reducing tumour size. Tumour size reduction and tumour growth stabilisation were described in 5 and 70% respectively of the 19 patients treated with high-dose lanreotide (up to 12 mg/day) in one study (Eriksson et al. 1997). In another study, in which 30 patients received s.c. injections of 5 mg lanreotide thrice a day for a period of 1 year, one complete and one partial remission in patients with functional midgut tumours were noticed; in the same study, 11 patients had stable disease (36%), while in another 11 patients the tumour progressed after 3–12 months of treatment (Faiss et al. 1999). Interestingly, in a study in which tumour biopsy specimens taken before and during SSA treatment were assessed, treatment with high-dose SSAs induced apoptosis in NETs, while this was not observed during treatment with low-dose SST (Imam et al. 1997). Accordingly, it is still hard to predict which patient will respond to which treatment dosage in terms of tumour growth inhibition.
The effect of SSA administration in combination with interferon on tumour growth has been assessed in a few studies, with variable results. One study indicated that this combination seemed to reduce the tumour progression compared with octreotide treatment alone, but the effect on 5-year survival was not significant (Kolby et al. 2003). Another study suggested that the addition of α-interferon to octreotide showed anti-proliferative efficacy in a subgroup of patients with advanced metastatic disease unresponsive to octreotide monotherapy, and prolonged survival was reported in the responder group (Frank et al. 1999). However, most published data do not support a major effect of interferons over and above that of SSAs. In hindgut carcinoid tumours, which are usually not associated with a clinical syndrome, there are no studies regarding the effect of SSAs on tumour growth in these patients.
In a study of 15 patients diagnosed with malignant gastrinoma (Shojamanesh et al. 2002), treatment with octreotide LAR had an anti-proliferative effect in about 50% of these patients, including one patient with tumour regression and another seven patients with tumour stabilisation (for a mean period of 25 months). Interestingly, patients with slow-growing tumours were more likely to respond to this treatment, and the authors therefore recommended that octreotide treatment should replace chemotherapy as the standard treatment for these patients. The high expression of SSTRs on gastrinomas has been considered as an opportunity to administer radiolabelled SSAs, in order to achieve a cytotoxic effect (111In-labelled analogues, 90yttrium or 177lutetium; Jensen 2004).
In a prospective multicentre trial including 103 metastatic GEP-NETs patients, in which 15 patients were diagnosed with non-functional pancreatic tumours, the octreotide effect on tumour growth after 1 year of treatment was investigated (Arnold et al. 1996); while tumour growth stabilised in only three patients from this subgroup, it progressed in another eight patients. Recently, a case report described that octreotide LAR was useful in achieving tumour regression in one and in preventing tumour progression in another patient diagnosed with a metastatic non-functioning neuroendocrine pancreatic tumour (Koehler et al. 2008). At least one large international study is currently underway to answer the question as to whether SSAs are of value in preventing tumour progression is such asymptomatic patients.
Regarding the anti-proliferative effects of SSAs in the treatment of other NETs, the literature is scanty. In a study of MTCs, disease stabilisation was achieved in three patients and minor tumour regression in two out of the seven patients included (Vitale et al. 2000). Radionuclide therapy using SSAs has been used in MTC patients, which received 111In-octreotide or 90Y-lanreotide in the MAURITIUS trial (Virgolini et al. 2002); the initial results were encouraging, suggesting that more prospective studies are needed.
While the effect of octreotide in treating bronchial carcinoids is mainly symptomatic, its anti-tumour effect is controversial: in a study of 31 patients with metastatic pulmonary carcinoids (Granberg et al. 2001), SSAs given as single drug treatment were associated with progressive disease; the combination of α-interferon and octreotide stabilised tumour growth in only 15% of the cases. However, s.c. administration of octreotide at a daily dosage of 1500 μg induced reduction/complete resolution of the liver metastases in three out of the seven patients included in another study (Filosso et al. 2002).
Rarely, malignant phaeochromocytomas may be octreotide, but not metaiodbenzylguanidine, avid; in such cases, therapy with octreotide may be useful (van der Harst et al. 2001); occasionally, disease stabilisation has been reported in a few patients with malignant chromaffin cell tumours treated with radionuclide SSAs, such as 111In-octreotide and 90Y-octreotide (DOTATOC; Valkema et al. 2002).
New directions in the treatment of NETs
It has been shown that subtypes of SST and dopamine receptors may form homo- and heterodimers at the membrane level, and that this receptor ‘interconnection’ may be stimulated by addition of either dopamine or SST. Furthermore, the development of SSTR subtype-specific analogues or chimaeric analogues, binding to SSTR2, SSTR5 and dopamine 2 receptors, has demonstrated promising clinical results. Lately, a number of new molecules (subtype selective analogues and antagonists as well as bi-specific and hybrid SST/dopamine compounds), have been developed (Ferone et al. 2007). Their activity is heterogeneous, being studied in animal and human cell lines, and also in primary cultures from human tumours, but further studies are needed to understand their complex biological effects. Phase I studies are currently underway with a chimaeric SSTR/dopamine receptor agonist, dopastatin (Ipsen, Paris, France).
SSTR targeted radiotherapy (peptide receptor radionuclide therapy, PRRT) represents a new advance in the treatment of NETs, being based on the presence of SSTRs in a higher density on these tumours and on their ability to form a receptor-ligand complex, permitting the internalisation-accumulation process of the radiopharmaceutical inside the tumour (Kaltsas et al. 2004).
While initially PRRT was performed using indium-111, with limited results (Fjalling et al. 1996, Kwekkeboom et al. 2005), the development of radiometal labelling chelators, such as DOTA (1,4,7,10-tetrazacyclo-dodecane-tetraacetic acid), which may be combined with metal ions (such as gallium, yttrium or lutetium), has allowed new therapeutic applications (Heppeler et al. 2000). In a phase II study in which the tumour response to targeted irradiation therapy using the radiolabelled SSA 90Y-DOTATOC in 41 patients with GEP-NET and bronchial tumours was evaluated, the overall response rate was 24% (36% for endocrine pancreatic tumours; Waldherr et al. 2001). Complete remission was observed in only 2% (1 out of 41), partial remissions in 22% (9 out of 41), a minor response in 12% (5 out of 41), stable disease in 49% (20 out of 41) and progressive disease in 15% (6 out of 41). The median duration of response had not been reached at 26 months, and the 2-year survival time was 76 months. There was a significant symptomatic improvement in about 83% of the patients suffering from the malignant carcinoid syndrome and the treatment was well tolerated. In a study of 103 patients with NETs, mostly GEP tumours, promising results were obtained when 177Lu-octreotate was used (Kwekkeboom et al. 2005): complete remission was reported in three patients (2%), partial remission in 32 patients (26%), a minor response in 24 patients (19%), stable disease in 44 patients (35%) and progressive disease in 22 patients (18%). High uptake on pretherapy SSTR imaging and a limited number of liver metastases were positively correlated with a higher remission rates, whereas progressive disease was significantly more frequent in patients with a low-performance score and extensive disease. Median time to progression in 103 patients who either had stable disease or tumour regression was more than 36 months. In a preclinical study using the combination of 90Y- and 177Lu-labelled analogues (de Jong et al. 2005) in animals bearing tumours of various sizes, combination treatment was superior to either 90Y or 177Lu analogues on their own in terms of the anti-tumour effects. In the near future, treatment with both 90Y and 177Lu coupled to a SSA will be subject to clinical trials, and available data suggest that 177lutetium may be more effective for smaller tumours whereas 90yttrium may be more effective for larger tumours (Oberg & Eriksson 2005, Chan & Kulke 2007). While PRRT appears to be a significant progress in the treatment of these tumours, there are many unresolved questions, such as which is the best time for its administration, or what is the most appropriate radioligand/combination to be used in an individual patient, and how the doses should be fractionated.
Conclusions
Data regarding the anti-proliferative mechanisms of SSAs and their role in the treatment of NETs are heterogeneous, and therefore difficult to analyse; this is complicated by the variability of the tumour types (in terms of site of origin, tumour histology or sites of metastases), use of different SSAs and of different dosages, lack of significant tumour progression before starting the treatment with SSAs, and the lack of randomised prospective studies. Although the SSAs have been shown to be very useful for the symptomatic and biochemical improvement in patients with NETs, specifically functional GEP tumours, their anti-proliferative effects are less impressive. In a study in which data from 62 published studies in this field were systematically analysed (Eriksson & Oberg 1999), stabilisation of tumour growth for a period of 8–16 months was observed in about 50% of the patients, while tumour regression was reported only in about 10–20% of patients. The mechanisms of these effects are equally obscure, but almost certainly include direct effects on proliferative signalling pathways, especially those involving MAPK, PI3K and Akt, activation of apoptosis, and effects on angiogenesis. A more detailed understanding of these mechanisms will almost certainly aid in the design of more effective analogues in terms of tumour regression as opposed to simple inhibition of secretion. Important directions for the use of SSAs in the future should include studies regarding their optimal dosage and modes of administration and the development of new slow release, SSTR subtype-specific compounds. The analysis of the SSTR status specifically for each patient, and studies of individual tumour biological behaviour, will help to optimise treatment and to add new insights into the mechanisms of action and the role of SSAs in the therapy of NETs.
Declaration of interest
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
- © 2008 Society for Endocrinology
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










