Von Hippel-Lindau disease and endocrine tumour susceptibility
- Section of Medical and Molecular Genetics and Cancer Research UK Renal Molecular Oncology Group, University of Birmingham, Institute of Biomedical Research, Birmingham B15 2TT, UK and West Midlands Regional Genetics Service, Birmingham Women’s Hospital, Birmingham B15 2TG, UK
- (Requests for offprints should be addressed to E R Maher at the first address; Email: E.R.Maher{at}bham.ac.uk)
Abstract
Von Hippel-Lindau (VHL) disease is a dominantly inherited familial cancer syndrome caused by mutations in the VHL tumour suppressor gene. VHL disease is characterised by marked phenotypic variability and the most common tumours are haemangioblastomas of the retina and central nervous system and clear cell renal cell carcinoma. However, endocrine tumours, most commonly phaeochromocytoma and non-secretory pancreatic islet cell cancers, demonstrate marked interfamilial variations in frequency and are significant causes of morbidity and, sometimes, mortality. Genotype–phenotype correlations have revealed that certain missense mutations are associated with a high risk of phaeochromocytoma but total loss of function mutations are associated with a low risk. Furthermore, rare mutations may predispose to a phaeochromocytoma-only phenotype. Germline VHL mutations may be detected in 5–11% of all phaeochromocytoma cases and mutation analysis of VHL and other phaeochromocytoma susceptibility genes (SDHB, SDHD and RET) should be performed in all cases of familial, multiple or early onset phaeochromocytomas, and considered in other cases. The VHL gene product has a key role in regulating the stability of hypoxia-inducible factors (HIF-1 and HIF-2) such that inactivation of VHL leads to up-regulation of HIF-1 and HIF-2 protein expression and activation of hypoxic gene response pathways. Germline SDHB and SDHD mutations also lead to increased expression of HIF target genes, but it appears that phaeochromocytoma susceptibility in VHL disease cannot be attributed to HIF activation alone. Recently, it has been suggested that an HIF-independent failure of developmental apoptosis is a common feature of all inherited phaeochromocytoma susceptibility syndromes.
Introduction
Although the first descriptions of von Hippel-Lindau (VHL) disease were in the 19th century, phaeochromocytoma was first associated with VHL disease only about 50 years ago (Glushien et al. 1953). Traditionally, an inherited cause of phaeochromocytoma has been estimated to account for ~10% of all cases but the identification of succinate dehydrogenase subunit mutations as a novel cause of familial phaeochromocytoma has prompted increased awareness and interest in the phaeochromocytoma susceptibility (Astuti et al. 2001a,b, Maher & Eng 2002, Neumann et al. 2002). We review the clinical features and molecular genetics of VHL disease with particular emphasis on the link between VHL disease and endocrine tumours.
Clinical phenotype of VHL disease
VHL disease has a birth incidence of ~1 in 36 000 and ~20% of cases arise as de novo mutations without a family history (Maher et al. 1991, Richards et al. 1995). VHL disease most commonly presents with retinal or cerebellar haemangioblastomas. The third major feature is clear cell renal cell carcinoma, and cystic disease affecting the kidneys, pancreas and epididymis is also frequent. Phaeochromocytoma is a notable feature because of marked interfamilial variation such that although the overall frequency is 10–20%, in many families it is absent while in other kindreds it is the most frequent manifestation of VHL disease (Maher 2004). Pancreatic islet cell tumour is the other major endocrine tumour seen in VHL disease although occasionally hyperparathyroidism and carcinoid have been reported (Binkovitz et al. 1990, Fellows et al. 1990, Hough et al. 1994, Arao et al. 2002, authors’ unpublished observations). Non-endocrine features of VHL disease have been reviewed in detail elsewhere (Maher 2004). The three major features are retinal angiomas (mean age at diagnosis 25 years), central nervous system (CNS) haemangioblastomas (cerebellum (38%, mean age 29 years), spinal cord (58%, mean age 34 years), brainstem (10%) and rarely supratentorial) and clear cell renal cell carcinoma (RCC) (mean age 35–40 years) (see Maher 2004 and references within). In most cases the lifetime risk for developing retinal angioma, CNS haemangioblastoma or clear cell RCC is ~70%, but the precise risk varies according to the nature of the germline mutation (see later). Endolymphatic sac tumours have only been recognised as a complication of VHL disease in the last 10 years but have been reported to occur in up to 11% of patients (Manski et al. 1997).
The mean age at diagnosis of phaeochromocytoma in VHL disease is ~20 years compared with 43.9 years in sporadic cases, reflecting both increased detection through surveillance (see below) and a predisposition to early onset tumours (Maher et al. 1990, Neumann et al. 2002). Phaeochromocytoma in VHL disease is usually intra-adrenal but ~10% are extra-adrenal (Neumann et al. 1993, Walther et al. 1999). Many VHL phaeochromocytomas are asymptomatic at diagnosis and while this is, in part, related to the early diagnosis by routine screening the number of asymptomatic tumours is higher than in other familial disorders such as multiple endocrine neoplasia Type 2 (MEN2). Compared with MEN2, VHL phaeochromocytomas are relatively less likely to express the enzyme phenylethanolamine-N-methyltransferase (PNMT) that converts noradrenaline to adrenaline such that they almost exclusively produce nor-adrenaline. However, in MEN2 there is a relative excess of adrenaline that causes the characteristic clinical manifestations such as tachycardia and episodes of tremulousness (Eisenhofer et al. 2001).
The most frequent pancreatic manifestation of VHL disease is benign serous cysts that occur in up to 90% of VHL patients and rarely cause clinical disease (Neumann et al. 1991, Hough et al. 1994, Hammel 2000). Benign microcystic adenomas also occur in a minority of cases (~12%) (Hammel et al. 2000). More significantly, 5–10% of VHL patients develop pancreatic tumours, most commonly non-secretory islet cell tumours (also known as neuro-endocrine tumours (NET)) which may be multifocal in 50% of affected individuals (Lubensky et al. 1998, Libutti et al. 1998, 2000, Marcos et al. 2002). These tumours are often asymptomatic and are usually detected incidentally during routine abdominal surveillance of VHL patients (Hough et al. 1994, Libutti et al. 1998, Hammel et al. 2000). The mean age at diagnosis of pancreatic NETs in VHL patients is younger than in sporadic cases (35 vs 58 years) (Lubensky et al. 1998). This earlier diagnosis may also contribute to the better prognosis of VHL-associated compared with sporadic NETs. Most VHL NETs are less than 3 cm and are slow growing. Although fewer than 10% metastasise, larger lesions may be more sinister and Marcos et al. (2002) found that ~20% of lesions >3.0 cm in diameter had metastasised to the liver.
Molecular genetics of VHL disease
The VHL tumour suppressor gene comprises 3 exons and encodes two proteins, a full length 213 amino acid protein, which migrates with an apparent molecular weight of ~28–30 kDa and a shorter 160 amino acid protein translated from an internal translation initiation site at codon 54. The 4.7 kb mRNA is widely expressed in both fetal and adult tissues, thus differential tissue expression does not account for the tissue-specific manifestations of VHL disease (Latif et al. 1993, Richards et al. 1996). Germline VHL mutations have been characterised in >500 patients and have provided a wealth of data for genotype–phenotype correlations (see later). VHL mutations may cause (a) VHL disease, (b) isolated familial phaeochromocytoma (Type 2C VHL disease) and (c) autosomal recessively inherited polycythaemia due to homozygous missense mutations (e.g. c.C811T, R200w in Chuvash polycythaemia) (Latif et al. 1993, Crossey et al. 1994, 1995, Chen et al. 1995, Neumann et al. 1995, Zbar et al. 1996, Woodward et al. 1997, Ang et al. 2002, Pastore et al. 2003). Among patients with VHL disease, ~40% of mutations are genomic deletions and the rest are predominantly truncating or missense mutations (which do not occur within the first 53 amino acids). Most intragenic mutations are restricted to a small number of families but notable exceptions are recurrent missense mutations at a CpG mutation hotspot at codon 167 (e.g. p.R167W and p.R167Q) and the p.Y98H missense founder mutation that originated in South-West Germany (the ‘Black Forest’ mutation) (Brauch et al. 1995, Richards et al. 1995).
The phenotypic variability that is characteristic of VHL disease may reflect mosaicism or modifier effects, but allelic heterogeneity is the major cause (Crossey et al. 1994, Webster et al. 1998, Sgambati et al. 2000). Thus, there are complex genotype–phenotype correlations in VHL disease particularly with regard to the presence or absence of phaeochromocytoma. VHL disease has been divided into Type 1 kindreds (in which affected individuals may have retinal or CNS haemangioblastomas and RCC but not phaeochromocytoma) and Type 2 kindreds (at least one affected individual has phaeochromocytoma). Type 2 kindreds are further subdivided into Type 2A (in which retinal and CNS haemangioblastomas but rarely RCC occur) and Type 2B (haemangioblastomas, RCC and phaeochromocytoma occur) (Crossey et al. 1994, Brauch et al. 1995, Maher et al. 1996, Zbar et al. 1996). Subsequently, a subset of families with isolated familial phaeochromocytoma were shown to have germline VHL gene missense mutations (Type 2C VHL disease) (Crossey et al. 1995, Neumann et al. 1995, Woodward et al. 1997). Whilst in some cases this reflected variable expression (e.g. p.R167w mutations that are usually associated with a Type 2B phenotype were found), in other families the mutation had not been described in other subtypes and appeared to be associated with a phaeochromocytoma only phenotype. Most patients with Type 2 VHL disease have missense mutations whereas in Type 1 families large deletions and truncating mutations predominate (Crossey et al. 1994, Maher et al. 1996, Zbar et al. 1996). Thus, it appeared that complete loss of function mutations were associated with a low risk of phaeochromocytoma, implying that Type 2 missense mutations may retain some functional activity. Consistent with this hypothesis, it was subsequently demonstrated that many missense mutations causing a Type 1 phenotype involved core hydrophobic residues and were predicted to disrupt protein structure, whereas Type 2 phenotype missense mutations involved substitutions at a surface amino acid that does not cause a total loss of function (Stebbins et al. 1999). Although no specific genotype–phenotype correlations have been defined for pancreatic NETs occurring in VHL disease, it has been observed that they may be more frequently associated with phaeochromocytoma (Binkovitz et al. 1990, Marcos et al. 2002). Thus Marcos et al. (2002) reported that up to 40% of VHL patients with a pancreatic NET had a surgically confirmed adrenal phaeochromocytoma. Such an association may reflect their shared embryonic neural crest origins and common mechanisms of tumourigenesis.
Differential diagnosis of familial phaeochromocytoma
Phaeochromocytoma susceptibility is associated with VHL disease, multiple endocrine neoplasia Types 2A and 2B, neurofibromatosis Type 1 (NF1) and also succinate dehydrogenase (SDH) subunit mutations. Whilst the clinical features of MEN2 and NF1 are well known, SDH subunit mutations were recognised to cause susceptibility to phaeochromocytoma and head and neck paraganglioma relatively recently (Baysal et al. 2000, Grimm et al. 2000, Astuti et al. 2001a,b, Niemann & Muller 2000). Germline mutations in three (SDHB, SDHC and SDHD) of the four SDH subunits have been associated with inherited phaeochromocytoma, but germline mutations in SDHC are rare (Niemann & Muller 2000). In patients presenting with familial, childhood or multicentric phaeochromocytoma, 30–40% will have a germline VHL mutation and a similar proportion of germline SDHD or SDHB mutation (Woodward et al. 1997, Astuti et al. 2001b, authors’ unpublished observations). In an unselected series of phaeochromocytoma cases, ~25% had a germline VHL (11%), SDHB (5%), SDHD (4%) or RET (MEN2) (5%) mutation (Neumann et al. 2002). However, in other series the frequency has been lower (15–20%) although this is still significantly higher than the ‘classic’ 10% (Gimenez-Roqueplo et al. 2003, authors’ unpublished observations). Compared with patients with germline VHL or RET mutations, there is an increased frequency of extra-adrenal phaeochromocytomas (and head and neck paragangliomas) in individuals with SDHB and SDHD mutations, and an increased rate of malignancy in those with SDHB mutations. Although VHL disease, MEN2 and SDHB mutations show autosomal dominant transmission patterns (albeit with variable expression and age-dependent penetrance), SDHD mutations only cause disease after paternal transmission and an individual who inherits the mutation from their mother will be clinically unaffected (Baysal et al. 2000, Astuti et al. 2001a).
Mechanisms of tumourigenesis
The VHL tumour suppressor gene (TSG) has features of a classical retinoblastoma-like TSG in that all tumour types from VHL patients may demonstrate inactivation of the wild-type VHL allele by allele loss, mutation or methylation (Prowse et al. 1997). Furthermore, sporadic clear cell RCC and sporadic haemangioblastomas may demonstrate somatic inactivation and loss, resulting in biallelic VHL inactivation (Foster et al. 1994, Gnarra et al. 1994, Kanno et al. 1994). However, somatic VHL mutations are rare in phaeochromocytoma and, interestingly, somatic inactivation of RET, SDHB and SDHD is also infrequent in sporadic phaeochromocytomas (Eng et al. 1995, Astuti et al. 2001b, 2003).
VHL protein function
The identification of the VHL gene did not provide any immediate clues to the function of the VHL protein (pVHL) as the protein sequence did not resemble any known protein (Latif et al. 1993). However, pVHL was shown to form a tetrameric (VCBC) complex with elongins B and C and Cul2 (a member of the cullin family) (Pause et al. 1997, Lonergan et al. 1998). In addition, structural homologies were noted between the VCBC complex and the yeast SCF complex which regulates proteosomal degradation of protein targets specified by the F-box component. Thus, targeted proteins are tagged for degradation by the addition of a ubiquitin tail by a ubiquitin ligase (Lonergan et al. 1998). Moreover, the Rbx-1 protein (a key component of SCF complexes) was then shown to associate with the VCBC complex (Kamura et al. 1999).
VHL-related tumours are highly vascular and demonstrate over-production of hypoxia-inducible mRNAs such as vascular endothelial growth factor (VEGF) (Wizigmann-Voos et al. 1995). Many hypoxia-inducible mRNAs are under the control of heterodimeric transcription factors (HIF-1 and HIF-2), which consist of a degradable alpha subunit and a stable constitutively expressed beta subunit (Schofield & Ratcliffe 2004). Under normoxic conditions, HIF-1α and HIF-2α are rapidly polyubiquitylated and destroyed by the proteosome, but under hypoxic conditions the alpha subunits are stabilised and HIF-1 and HIF-2 activate transcription of a wide repertoire of hypoxia-inducible mRNAs (Schofield & Ratcliffe 2004 and references within).
The crucial link between pVHL and the regulation of hypoxia-inducible mRNAs was provided by Maxwell and colleagues who showed that pVHL interacts with the regulatory alpha subunit of HIF and targets it for oxygen-dependent polyubiquitylation and proteosomal degradation (Maxwell et al. 1999, Cockman et al. 2000). pVHL mutants failed to degrade the alpha subunits leading to stabilisation of HIF-1 and HIF-2 and inappropriate expression of hypoxia-inducible mRNAs (see Fig. 1⇓). The oxygen-dependent interaction of pVHL with HIF-α is provided by the hydroxylation status of key HIF-α proline residues (Pro-402 and Pro-564) (Ivan et al. 2001, Jaakkola et al. 2001, Masson et al. 2001, Yu et al. 2001). Thus, in the presence of oxygen, HIF-α subunits are hydroxylated at the conserved prolyl residues by members of the egg laying defective nine (EGLN) family (also known as the PHD family) (Bruick & McKnight 2001, Epstein et al. 2001). Molecular oxygen and 2-oxoglutarate are essential cosubstrates and iron is an essential cofactor (Epstein et al. 2001, Schofield & Ratcliffe 2004) and, in the absence of these, hydroxylation does not occur and pVHL is unable to bind the alpha subunits. In humans, three EGLN homologues have been implicated in HIF-α modification (PHD1/EglN2/HIFPH1, PHD2/EglN1/HIFPH2 and PHD3/EglN3/HIFPH3) (Bruick & McKnight 2001, Epstein et al. 2001).
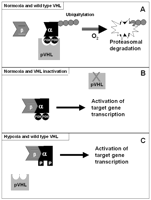
Relationship between VHL protein (pVHL) and regulation of hypoxia-inducible factor-α subunits. (A) Under normoxic conditions, pVHL binds to HIF-α subunits leading to ubiquitylation and proteosomal degradation. The ability of pVHL to bind an HIF-α subunit is dependent on hydroxylation of two proline residues (POH). (B) In a tumour with pVHL inactivation, the lack of wild-type pVHL allows HIF-α and HIF-β subunits to interact and the heterodimeric transcription factors activate expression of a wide repertoire of hypoxia-inducible genes. (C) In hypoxia, the two critical proline residues in the α subunit are not hydroxylated (P) and pVHL is unable to bind HIF-α, leading to activation of the hypoxia-inducible gene response.
pVHL has also been implicated in a variety of cellular processes including regulation of angiogenic factors and apoptosis, mRNA stability, fibronectin metabolism and microtubule stability (Ohh et al. 1998, Devarajan et al. 2001, Pioli & Rigby 2001, Hergovich et al. 2003). Whilst regulation of angiogenic factors is clearly an HIF-dependent pVHL function, for some other processes it has not been clear whether HIF-independent mechanisms were operating and to what extent HIF-dysregulation per se promotes tumourigenicity. However, over-expression of mutant HIF-2 protein resistant to pVHL-mediated proteolysis was found to override pVHL tumour suppressor activity and promote tumourigenesis of an RCC cell line (Kondo et al. 2002). However, in similar experiments HIF-1 over-expression did not promote tumourigenesis (Maranchie et al. 2002). It appears that HIF-1 and HIF-2 have overlapping but differing repertoires of targets genes and may have different roles in tumourigenesis (Kondo et al. 2002, Maranchie et al. 2002, Raval et al. 2005). Nevertheless, it seems that HIF-2 can have different effects on tumour growth in different tumour types (Acker et al. 2005).
The precise role of HIF dysregulation in the pathogenesis of VHL-related phaeochromocytomas is unclear. Thus both Type 2A and Type 2B VHL mutations (associated with a high risk of phaeochromocytoma) are unable to regulate HIF (Clifford et al. 2001) and comparison of gene expression patterns in VHL and MEN2 phaeochromocytomas revealed that many of the genes over-expressed in VHL compared with MEN2 tumours were linked to the hypoxia-driven angiogenic pathways (e.g. VEGF, placental growth factor, angiopoietin 2, tie-1, VEGF receptor 2 and neuropilin-1) (Eisenhofer et al. 2004). Phaeochromocytomas from SDHB and SDHD gene carriers demonstrate evidence of HIF activation and over-expression of hypoxia-inducible genes (Gimenez-Roqueplo et al. 2001, Pollard et al. 2005) and SDH inactivation has been shown in vitro to inhibit HIF-α prolyl hydroxylases leading to stabilisation and activation of HIF-1α, further implicating HIF dysregulation in phaeochromocytoma tumourigenesis (Selak et al. 2005). Furthermore, comparison of gene expression profiles in VHL-, SDHB- and SDHD-associated phaeochromocytomas demonstrated a shared transcription profile of reduced oxidoreductase and increased angiogenesis/hypoxia target genes (Dahia et al. 2005). However, the role of HIF-1 and HIF-2 in phaeochromocytoma tumourigenesis may be complex because Type 1 VHL mutations (with a low risk of phaeochromocytoma) are also unable to regulate HIF, and Type 2C (phaeochromocytoma only) pVHL mutants retain the ability to regulate HIF-α subunit degradation, suggesting that HIF dysregulation is not essential for the development of phaeochromocytomas in VHL disease (Clifford et al. 2001, Hoffman et al. 2001). Hence, it was suggested that the complex genotype–phenotype relationships observed in VHL disease were consistent with multiple and tissue-specific functions.
Recently, Lee et al. (2005) have suggested a novel HIF-independent pVHL function that links phaeochromocytoma in VHL disease to that in other familial phaeochromocytoma predisposition syndromes. Thus, they hypothesised that inherited phaeochromocytomas originate from sympathetic neuronal precursor cells that usually undergo c-Jun dependent apoptosis during embryogenesis when growth factors such as nerve growth factor (NGF) become limiting (Estus et al. 1994, Schlingensiepen et al. 1994, Ham et al. 1995, Xia et al. 1995) (see Fig. 2⇓). JunB is an antagonist of c-Jun such that increased levels of JunB attenuate c-Jun induced apoptosis in phaeochromocytoma cells with NGF withdrawal. Pathogenic NF1 and RET mutations are known to enhance signalling by NGF receptors and hence promote neuronal survival (Vogel et al. 1995, Dechant 2002). Lee et al. (2005) found that all pVHL mutants tested (including 2C mutants that retain the ability to degrade HIF) failed to down-regulate JunB following NGF withdrawal, thus promoting cell survival (the pVHL effect on JunB is mediated, in part, through an HIF-independent, atypical protein kinase C pathway). Furthermore, a similar effect was also demonstrated with activating mutations of RET. EglN3 (PHD3) (a member of the proline hydroxylase EGLN family) had previously been shown to be induced in sympathetic neurons following NGF withdrawl and to provoke apoptosis when over-expressed in phaeochromocytoma cells (Lipscomb et al. 1999, 2001, Straub et al. 2003) and Lee et al. (2005) reported (a) that EglN3, but not EglN1, acts downstream of c-Jun and is both necessary and sufficient to induce neuronal apoptosis following NGF withdrawal and (b) that EglN3 is sensitive to changes in SDH activity such that after SDH inactivation, succinate accumulation inhibits the activity of EglN3 thus preventing apoptosis of the neuronal precursor cells. Although SDH is a mitochondrial enzyme, the succinate that accumulates is transported to the cytosol by the dicarboxylate carrier located on the inner mitochondrial membrane. Thus, these findings provide a common link between phaeochromocytoma development in inherited phaeochromocytoma susceptibility syndromes as (a) NF1 inhibits downstream signalling by the NGF receptor, TrkA, and loss of NF1 promotes NGF independent survival of embryonic peripheral neurons (Vogel et al. 1995) and (b) activation of RET, like loss of pVHL, has been shown to lead to the induction of JunB and thus attenuates apoptosis after NGF withdrawal. In addition, the concept that germline NF1, RET, SDH subunit and VHL mutations promote phaeochromocytoma development by allowing sympathetic neuronal progenitors to escape from developmental apoptosis is consistent with the observation that somatic inactivation of these genes is infrequent in sporadic phaeochromocytoma (whereas somatic VHL inactivation occurs in most sporadic clear cell RCC) (Maher & Eng 2002). It should be noted that whereas all pVHL mutants fail to down-regulate JunB (including Type 1 mutants which are not associated with phaeochromocytoma), pVHL has multiple effects on EglN3, and Type 1 and Type 2 pVHL mutants differ with respect to their effect on EglN3/PHD3 expression (increased in Type 1 mutants). Thus, Type 1 mutations increase EglN3 expression whereas Type 2 mutations result in reduced EglN3 expression.
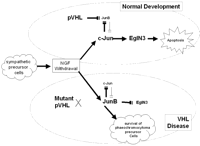
Model of phaeochromocytoma in von Hippel-Lindau disease (Lee et al. 2005). (Top panel) In normal development as nerve growth factor (NGF) levels become limiting, sympathetic neuronal precursor cells proceed to apoptosis via a cJun/EglN3 pathway. Whilst the pro-apoptotic effect of cJun is antagonised by JunB, pVHL inhibits JunB expression so that apoptosis is favoured. (Lower panel) In the presence of a Type 2 pVHL mutant, the failure of pVHL-induced repression of JunB anatagonises cJun and EglN3 such that some sympathetic neuronal precursor cells fail to undergo apoptosis and may develop into a phaeochromocytoma.
Clinical and molecular investigations of VHL disease and other hereditary phaeochromocytoma syndromes have provided new insights into mechanisms of cellular oxygen sensing and the role of HIF in the molecular pathogenesis of VHL-related tumours. However, analysis of genotype–phenotype correlations in VHL disease indicated the involvement of HIF-independent pathways in phaeochromocytoma development. The elucidation of the role of NGF/JunB/EglN3-related pathways in developmental apoptosis has provided novel insights into mechanisms of tumourigenesis in familial and sporadic phaeochromocytoma.
Acknowledgments
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
- © 2006 Society for Endocrinology










