Emerging role of the endothelin axis in ovarian tumor progression
- Molecular Pathology and Ultrastructure Laboratory, Regina Elena Cancer Institute, Via delle Messi d’Oro 156, 00158 Rome, Italy
- (Requests for offprints should be addressed to A Bagnato; Email: bagnato{at}ifo.it)
Abstract
Ovarian cancer is the leading cause of gynecologic cancer-related deaths. The endothelin (ET) axis, which includes ET-1, ET-2, ET-3, and the ET receptors, ETAR and ETBR, represents a novel target in tumor treatment. ET-1 may directly contribute to tumor growth and indirectly modulate tumor–host interactions in various tumors such as prostatic, ovarian, renal, pulmonary, colorectal, cervical, breast carcinoma, Kaposi’s sarcoma, brain tumors and melanoma. Extensive experimental evidence links ETAR overexpression with tumor progression in ovarian cancer. ETAR engagement can in fact activate multiple signal transduction pathways including protein kinase C, phosphati-dylinositol 3-kinase, mitogen-activated protein kinase and transactivate epidermal growth factor receptor, which play a role in ovarian tumor growth and invasion. The effects of ETAR signaling are wide ranging and involve both cancer cells and their surrounding stroma, including the vasculature. Upon being activated, the ETAR mediates multiple tumor-promoting activities, including enhanced cell proliferation, escape from apoptosis, angiogenesis, epithelial–mesenchymal transition and increased motility and invasiveness. These findings indicate that activation of ETAR by ET-1 is a key mechanism in the cellular signaling network promoting ovarian cancer growth and progression. The predominant role played by ETAR in cancer has led to the development of small molecules that antagonize the binding of ET-1 to ETAR. The emerging preclinical data presented here provide a rationale for the clinical evaluation of these molecules in which targeting the related signaling cascade via ETAR blockade may be advantageous in the treatment of advanced stage ovarian carcinoma.
Introduction
Ovarian cancer, the leading cause of death from gynecological malignancy, is a highly metastatic disease characterized by widespread peritoneal dissemination and ascites (Agarwal & Kaye 2003). As the treatment of patients in advanced stages is still associated by low survival rates, the development of new treatment protocols depends on improved knowledge of the molecular mechanisms controlling tumor progression (Naora & Montell 2005). Cancer invasion is a state that emerges from a tumor–host microenvironment in which the host participates in the induction, selection and expansion of the neoplastic cells. During malignancy, tissue architecture, which is normally maintained by basement membrane delineation of tissue boundaries and cell–cell communication, is disrupted. In the microecology of the tumor–host invasion field, growth factor exchange between the participating cells stimulates migration, invasiveness, neovascularization and promotes proliferation and survival. Among these, endothelins are an example of such mediators. The endothelins, include three 21-amino acid (aa) peptides endothelin (ET)-1, ET-2 and ET-3, are widely distributed in tissues, and are produced by endothelial cells and many epithelial cell types (Levin 1995, Masaki 2004). ET-1, derived from vascular endothelial cells with potent vasoconstrictor activity, is encoded by a distinct gene and is regulated at the level of mRNA transcription (Yanagisawa et al. 1988). The primary translation product of the ET-1 gene is the 212-aa prepro-ET-1, which is cleaved by an endopeptidase to form the 38-aa big ET-1. The biologically active ET-1 is formed by endothelin-converting enzyme (ECE), an enzyme with intra-cellular and membrane bound isoforms (Xu et al. 1994). The half-life of ET-1 in the circulation is 7 min (Rubin & Levin 1994). Two pathways have been described for the clearance of endothelin: ETB receptor-mediated uptake followed by lysosomal degradation (Burkhardt et al. 2000, Bremnes et al. 2000) and catabolism by extracellular neutral endopeptidase (NEP). ET-1 production is stimulated by a variety of cytokines and growth factors, including IL-1α, TNF-α, TGF-β, PDGF, vasopressin, hypoxia and shear stress. Inhibitory factors include nitric oxide, prostacyclin and atrial natriuretic peptide (Rubin & Levin 1994). Endothelins exert their effects by binding to two distinct cell surface ET receptors, ETA and ETB. The ETB receptor (ETBR) binds the three peptide isotypes with equal affinity. In contrast, ETAR binds ET-1 with higher affinity than the other isoforms. Both receptors belong to the G protein-coupled receptor (GPCR) family, mediating pleiotropic actions of ETs and are distributed in a variety of cells and tissues in different proportions, suggesting the potential of opposing regulatory functions (Levin 1995, Masaki 2004). ET-1, has emerged as an important peptide in a host of biological functions, including development, cellular proliferation, apoptosis and cancer (Nelson et al. 2003). ET-1 has been implicated in the pathophysiology of a wide range of human tumors, including ovarian carcinoma. There has been a number of excellent reviews on the role of the ET-1 axis in tumors (Grant et al. 2003, Guise et al. 2003, Nelson et al. 2003, Bagnato & Natali 2004, Grimshaw 2005). This review will therefore focus on the role of ET-1 axis in ovarian tumor progression.
Expression of ET-1 axis in ovarian carcinoma
More than 90% of primary and 100% of metastatic ovarian cancers express ET-1 mRNA as detected by reverse-transcription-PCR and northern blot analysis. The ET-1 mRNA expression was significantly higher in tumors than in normal ovarian tissue. ETAR mRNA was also detected in 84% of the carcinoma examined, whereas ETBR mRNA was expressed in 40% of the tumors. The in vivo presence of ET-1 and ETAR protein was confirmed by immunohistochemistry, demonstrating a higher expression in primary and metastatic cancer cells. Radioligand binding studies showed that ET-1 producing cells also expressed functional ETAR, whereas no specific ETBR could be demonstrated (Bagnato et al. 1999). Ascites fluid in ovarian cancer patients provides a window on the cellular microenvironment of the tumor, as well as on growth factors that are produced by tumor cells. High levels of ET-1 were detected in the majority of ascitic fluids of patients with ovarian carcinoma, and were significantly correlated with vascular endothelial growth factor (VEGF) concentration. These results implicate that this molecule is relevant in the progression of ovarian carcinoma (Salani et al. 2000a). The increased levels of ET-1 in ascites fluid indicated that it could be a diagnostic marker, a prognostic indicator, or an indicator of response to therapy in malignancy.
Signaling pathways activated by ET-1
In ovarian tumor cells, ET-1 acts as a selective autocrine factor for ETAR. Engagement of ETAR leads to activation of a pertussis toxin-insensitive G protein that stimulates phospholipase C activity and increases intracellular Ca2+ levels, activation of protein kinase C, and mitogen activated protein kinase (MAPK). Cross-talk between cell surface receptors, which has been recognized as the mechanism capable of expanding the cellular communication signaling network, is now receiving further interest. Receptor cross-talk can, in fact, also occur among distinct families of receptors such as tyrosine kinase receptors and GPCR. In this context, we have previously shown that in ovarian cancer cells, ET-1 causes epidermal growth factor (EGF) receptor transactivation that is in part responsible for MAPK activation by a ligand-dependent mechanism involving a non-receptor tyrosine kinase, such as Src. This event leads, through the formation of Shc/Grb-2 complexes, to activation of the Ras/MAPK pathway (Bagnato et al. 1997, Bagnato & Catt 1998, Vacca et al. 2000, Nelson et al. 2003). ET-1 binding to the ETAR results in p125 focal adhesion kinase and paxillin activation, which are thought to transduce signals involved in tumor cell invasion. Furthermore, ET-1 triggers the activation of anti-apoptotic signaling through phosphatidylinositol 3-kinase (PI3-K)-mediated Akt pathways (Del Bufalo et al. 2002). These findings demonstrate the existence of multiple signal transduction pathways downstream to ETAR activation in ET-1 stimulated ovarian cancer cells (Fig. 1⇓).
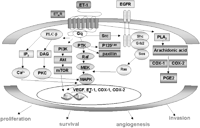
ET-1 induced signal transduction pathways in ovarian cancer cells. Binding of ET-1 to ETA receptor (ETAR) triggers signal transduction pathways through a pertussin-insensitive G protein that is coupled to the ETAR intracellular domain. Activation of phospholipase C (PLC), protein tyrosine kinases (PTKs; such as FAK and paxillin) ultimately results in the activation of the RAF/MEK/MAPK pathway. ET-1 also causes Src-mediated epidermal growth factor receptor (EGFR) transactivation that is in part responsible for MAPK activation. ET-1 also stimulates phosphatidylinositol 3-kinase (PI3-K)-mediated Akt activation. Parallel mobilization of intracellular calcium (Ca2+), activation of protein kinase C (PKC), MAPK and Akt induces nuclear transcription of genes, such as VEGF, ET-1, COX-1 and -2, leading to proliferation, survival, angiogenesis and invasion. Further analysis showed that ET-1 promotes cyclooxygenase (Cox)-1 and -2 expression and, in turn, prostaglandin (PG) E2 production, amplifying ET-1 driven VEGF production. DG, Diacylglycerol; IP3, inositol 1,4,5 triphosphate; MAPK, mitogen activated protein kinase; mTOR, mammalian target of rapamycin; MEK, MAPK kinase; p125 FAK, focal adhesion kinase; PLA, phospholypase A; VEGF, vascular endothelial growth factor.
Endothelin axis in tumor transformation and survival
The ET-1/ETAR autocrine pathway has been implicated in ovarian cancer progression, acting via a wide range of cancer relevant processes, such as cell proliferation, inhibition of apoptosis, angiogenesis, migration, and invasiveness. The demonstration of ET-1 as an important mediator in ovarian tumor progression clearly identifies the ETAR as a potential therapeutic target. This has propelled the development of several potent and selective ETAR receptor antagonists. These small molecules have contributed to our understanding of the physiopathological relevance of the ET-1 system and the beginning of translation of this information into clinical trials (Nelson et al. 2003).
Regulation of tumor cell growth
ET-1 activates various kinases that are known to be involved in cell proliferation, inducing rapid induction of early response genes including c-fos, c-jun and c-myc (Bagnato et al. 1997). ET-1 stimulates DNA synthesis and cell proliferation in various cell types, including VSMC, osteoblasts, glomerular mesangial cells, fibroblasts and melanocytes as well as different tumor cell types including ovarian cancer cells. In primary cultures and established ovarian carcinoma cell lines, spontaneous growth was significantly inhibited in the presence of BQ 123, a peptidic ETAR antagonist. In contrast, the ETBR antagonist, BQ 788, lacked this activity demonstrating that endogenous ET-1 acts as an autocrine modulator of ovarian carcinoma cell proliferation only through the ETAR (Bagnato et al. 1999). The mitogenic activity of ET-1 can be amplified by synergistic interactions with other growth factors including EGF, basic fibroblast growth factor (bFGF), insulin, insulin-like growth factor (IGF), platelet-derived growth factor (PDGF), transforming growth factor β(TGFβ), and interleukin-6 (IL-6) (Battistini et al. 1993). In ovarian cancer cells, ET-1 was found to stimulate DNA synthesis with the same efficacy as EGF, and at maximally effective concentrations its action was additive to that of EGF. The findings that EGFR transactivation is in part responsible for the mitogenic effect of ET-1/ETAR pathway and that ET-1 exerts additive proliferative effects in the presence of EGF, suggest that the coexistence of ET-1 and EGF autocrine circuits in tumor cells could provide maximal growth advantage (Bagnato et al. 1997, Vacca et al. 2000).
Regulation of apoptosis
ET-1 is an anti-apoptotic factor in different cell types, indicating that the peptide may also modulate cell survival pathways. In ovarian carcinoma cells, the addition of ET-1 markedly inhibited serum withdrawal and paclitaxel-induced apoptosis in a concentration-dependent manner. Paclitaxel-induced apoptosis resulted in the phosphorylation of Bcl-2 that was suppressed by the addition of ET-1. Further analysis of the survival pathway demonstrated that ET-1 stimulated Akt activation was dependent on PI3-K. Interestingly, the addition of a specific ETAR antagonist blocked the ET-1 induced resistance to paclitaxel-mediated apoptosis, indicating that ET-1 contributes to paclitaxel resistance through ETAR binding via activation of anti-apoptotic signaling pathways, such as Akt. Specific ETAR antagonists may therefore provide an additional approach to the treatment of ovarian carcinoma, in which ETAR blockade could result in tumor inhibition by reducing tumor growth and by inducing apoptosis. Furthermore, when combined with the conventional chemotherapy the ETAR antagonists would more effectively induce apoptosis by contributing to the reversal of paclitaxel resistance (Del Bufalo et al. 2002).
Endothelin axis in tumor progression and metastasis
Metastasis relapses remain a major challenge in ovarian cancer management. Tumor metastasis involves the invasion of the tumor into surrounding tissues, with dissemination and growth at distant sites. Factors involved in tumor progression include changes in cell adhesion, cell communication, increased migration or motility, invasiveness and angiogenesis. ET-1 has been shown to contribute to all of these processes, as discussed in the subsequent sections.
Regulation of tumor angiogenesis
Angiogenesis, the formation of new vessels from existing vasculature, is an important early event in tumor progression that begins in premalignant lesions, where this process is more accurately defined as a combination of angiogenesis and vasculogenesis. Initiation of angiogenesis is controlled by different regulators including local hypoxia, which activates the expression of angiogenic factors that can stimulate endothelial cell growth (Carmeliet & Jain 2000). Some tumor cells are able to form de novo extracellular matrix (ECM)-rich vascular channels expressing vascular-associated molecules, a phenomenon called vasculogenic mimicry (Folberg et al. 2000).
VEGF and bFGF are the principal regulators of neovascularization. Although growth factors, such as VEGF, and their cognate tyrosine kinase receptors are the best characterized mediators of angiogenesis, several GPCRs also have a role in angiogenesis. Among these GPCR agonists, we will discuss the role of ET-1. Endothelial cells actively produce and secrete ET-1 and simultaneously express ETBR as the major receptor population, indicating a potential autocrine role for endogenous ET-1. Moreover, ET-1 acting via positive autocrine feedback on ETBR, increases its own synthesis in human umbilical vascular endothelial cells (HUVEC). Previous studies demonstrated that ET-1 and ET-3, have dose-dependent stimulatory, proliferative and migratory effects, and accelerate wound healing on endothelial cells isolated from bovine adrenal capillaries and HUVEC. These findings suggested that ET-1 might exert angiogenic activity (Bagnato & Spinella 2002). During the formation of new blood vessels, endothelial cells were stimulated to release proteases, such as MMP-2 to migrate, proliferate and invade surrounding tissue to form capillaries. ET-1, similar to VEGF, induced these angiogenic effects in vitro and in vivo and in concert with VEGF, displayed a potent additive effect on the different stages of the angiogenic process. ET-1 signaling was mediated mainly by the ETBR, because addition of the selective ETBR antagonist, BQ 788, strongly inhibited the stimulatory activity of ET-1 (Salani et al. 2000b). Although ET-1 can directly modulate the different steps of angiogenesis, it can also act indirectly through the induction of major angiogenic factors, such as VEGF. Recent studies exploring a potential interaction between VEGF and ET-1 demonstrated that VEGF enhanced ET-1 mRNA expression and ET-1 secretion in endothelial cells. Similarly, in VSMC, ET-1, acting predominantly through the ETA receptor, enhanced VEGF mRNA expression and VEGF secretion, and stimulated VEGF-induced endothelial cell proliferation and invasion (Bagnato & Spinella 2002). This indicates that VEGF and ET-1 have reciprocal stimulatory interactions that result in concomitant proliferation of endothelial cells and VSMC. Furthermore, VEGF is involved in ET-1 mediated angiogenesis in chorioal-lantoic membrane (CAM) as demonstrated by the ability of a specific inhibitor of VEGF receptor activity to prevent ET-1 induced nodule formation and CAM neovascularization (Cruz et al. 2001). As VEGF and ET-1 might be up-regulated by various stimuli, including hypoxia, it is reasonable to propose that in tumor tissues, acute or chronic hypoxia might stimulate VEGF production through both direct and indirect effects, the latter also involving ET-1 secretion (Bagnato & Spinella 2002). Elevated expression of ET-1 and its cognate receptors is significantly associated with expression of VEGF and its receptors (KDR and flt-1) and tumor-induced vascularization, which was quantified as microvessel density (MVD), using antibodies against CD31, a specific marker of endothelial cells in ovarian carcinomas (Salani et al. 2000a). This suggests that ET-1 and VEGF might have complementary and co-ordinated role during neovascularization in this tumor. Thus, in ovarian carcinoma cell lines, ET-1 through the ETAR increases VEGF mRNA expression, inducing VEGF levels in a time-and dose-dependent fashion, and does so to a greater extent during hypoxia (Salani et al. 2000a). Transcriptional upregulation has an important role in the induction of VEGF expression and this has been linked to a critical mediator of hypoxia signaling, the hypoxia-inducible factor 1α (HIF-1α) (Forsythe et al. 1996). ET-1 promotes VEGF production through HIF-1α.
Analysis of HIF-1α protein stability showed that its degradation was reduced in ET-1 treated ovarian carcinoma cells compared with controls under both hypoxic and normoxic conditions, indicating that the induction of HIF-1α protein production by ET-1 is due to enhanced HIF-1α stability. After ETAR activation by ET-1, HIF-1α protein levels are increased, the HIF-1 transcription complex is formed and binds to the HRE binding site. Therefore, ET-1/ETAR induced HIF-1 accumulation in ovarian carcinoma cell lines might be responsible for increasing VEGF mediated angiogenesis (Spinella et al. 2002). Invasive tumor cells, including those of melanoma, prostate, breast and ovarian carcinomas, have been shown to form vasculogenic mimicry (Folberg et al. 2000). Sood et al. (2001) demonstrated that tumor cell-lined vasculature is exhibited by aggressive, but not by normal, ovarian surface epithelial cells. Thirty percent of human ovarian cancers have some degree of tumor cell-lined vasculature that is associated with advanced stage, high tumor grade, development of distant metastasis and poor overall survival (Sood et al. 2002, 2004). MMP-2 and MT1-MMP appear to play a key role in the development of vasculogenic-like networks and matrix remodeling by aggressive ovarian cancer cells. In addition, human ovarian cancers with MMP overexpression are more likely to have tumor cell-lined vasculature (Sood et al. 2004). Interestingly, the invasive ovarian cancer cells that are capable of generating tubular networks in vitro express both MMPs and ETAR, and produce ET-1. Furthermore ETAR antagonist treatment prevented the formation of tumor-lined vascular channels, generated by vasculogenic mimicry (Bagnato et al. unpublished results). Therefore, ETAR blockade treatment could also exert anti-angiogenic effect by acting on microvascular channels lined by tumor cells that overexpress ETAR, in the absence of endothelial cells expressing ETBR. Prostaglandins (PG) and their rate-limiting enzymes cyclooxygenase (COX)-1 and -2 are involved in the onset and progression of a variety of malignancies (Dannenberg & Subbaramaiah 2003). Moreover high COX-1 and -2 expression has been reported in association with elevated levels of proangiogenic factors in ovarian cancer (Denkert et al. 2002, Erkinheimo et al. 2004, Ferrandina et al. 2002, Gupta et al. 2003). The role of COX-1 on ovarian cancer progression has been highlighted by recent findings showing that COX-1 is the dominant pathway responsible for generating prostaglandins, and represents a potential target for the prevention and treatment of human ovarian cancer (Daikoku et al. 2005). In ovarian carcinoma cells, ET-1 significantly increases the expression of COX-1 and -2, at mRNA and protein levels, COX-2 promoter activity and PGE2 production. These effects depend on ETAR activation and involve multiple MAPK signal pathways, including p42/44 MAPK, p38 MAPK and transactivation of the EGFR (Spinella et al. 2004b).
In ovarian carcinoma, ET-1 by binding with ETAR induces PGE2 production, as the more expressed PG type, and increases the expression of PGE2 receptor type 2 (EP2) and type 4 (EP4) (Spinella et al. 2004a). There is increasing evidence that PGE2 contributes to tumor progression by promoting angiogenesis and that this effect is mediated by VEGF. COX-1 and -2 inhibitors blocked ET-1 induced PGE2 and VEGF release, demonstrating that both enzymes, although to a different extent, participates in PGE2 and VEGF production. The use of EP agonists and antagonists indicates that ET-1 and PGE2 stimulate VEGF production, principally through EP2 and EP4 receptors. At the mechanistic level, we found that the induction of PGE2 and VEGF involve ETAR activation and Src-mediated EGFR transactivation in ovarian carcinoma cells (Spinella et al. 2004a). These results indicate that impairing COX-1 and -2 and their downstream effects by targeting ETAR can be therapeutically advantageous (Spinella et al. 2004b), consistent with the association of elevated COX-2 levels with tumor progression and chemoresistance (Ferrandina et al. 2002).
Regulation of tumor invasiveness
As previously mentioned, high levels of ET-1 are present in the majority of ascitic fluids of ovarian cancer patients and are significantly correlated with VEGF ascitic concentrations, suggesting that ET-1 enhances the secretion of extracellular matrix-degrading proteinases and metastasis (Salani et al. 2000a). Thus, ET-1 acting through the ETAR consistently induced the activity of two families of metastasis-related proteinases, the matrix metalloproteinases (MMPs) and the urokinase type plasminogen activator system at several levels: mRNA transcription, zymogen secretion and proenzymes activation. ET-1, in fact, activates MMP-2, MMP-9, MMP-3, MMP-7 and MMP-13. In addition to soluble MMPs, ET-1 enhances the activation of membrane type 1-MMP (MT1-MMP) and the secretion of tissue inibitor of MMP (TIMP-1 and -2), increasing the net MMP/TIMP balance and gelatinolytic activity that causes rapid degradation of the ECM. In ovarian carcinoma cells, co-induction of uPA system by the concomitant stimulation of production and secretion of uPA and uPAR, and MMPs by ET-1 caused the highest invasive potential of tumor cells through the Matrigel (Rosanò et al. 2003b). ET-1 induced expression of COX-1/-2, and PGE2 amplified the ET-1 mediated effects on MMP activity and cell migration through a Matrigel layer, contributing to the invasive and migratory capability of ovarian carcinoma cells. EP2 and EP4 receptor antagonists blocked MMP activity and cell invasion, demonstrating that these receptors are the principal PGE2 receptor involved in these processes (Spinella et al. 2004a). In addition to direct activation of COX-1, -2 and PGE2, as well as MMP activity and cell invasion, ET-1 may induce these effects indirectly through interactions with EGFR. In this regard, ETAR-induced EGFR transactivation may serve as a prototype of inter-receptor signaling since multiple, apparently independent, pathways are coactivated by this network, resulting in MMP activation and invasiveness (Spinella et al. 2004a,b). Interestingly, the addition of an ETAR antagonist blocked ET-1-induced proteinase activation and tumor cell migration and invasion. Furthermore, in these cells ET-1 stimulated FAK and paxillin phosphorylation through the ETAR (Bagnato et al. 1997) which directly correlated with tumor cell migration and invasion. This indicates that ETAR antagonist can inhibit cell migration and possibly other FAK-associated processes which also contribute to invasion and metastasis by this tumor (Rosanò et al. 2001).
Regulation of intercellular communication
Following malignant transformation, stepwise changes in intercellular communication enable tumor cells to escape microenvironmental control from the normal surrounding tissue, thus promoting local invasiveness and metastatic spread. Human ovarian surface epithelial cells exhibit extensive gap junction intercellular communications (GJIC) and expression of different types of connexin (Cx), predominantly Cx43. Defects in intercellular communication, including reduced or inappropriate expression of Cx43, have emerged as key factors in ovarian carcinoma progression (Umhauer et al. 2000). In ovarian carcinoma cells, ET-1 via the ETAR induces transient and a dose-dependent reduction of GJIC (50–75%) and phosphorylation of Cx43 through the Src tyrosine kinase pathway, indicating that ET-1 promotes cellular uncoupling at the level of connexin maturation and subsequent degradation (Spinella et al. 2003). The capacity of ET-1 to disrupt gap junctions could serve as a basis to further evaluate the cell to cell metabolic uncoupling and cell detachment that occurs during tumor progression. This underlines the overall relevance of ETAR in regulating the complex array of cell–cell or cell–matrix interactions that promote ovarian carcinoma growth (Spinella et al. 2003).
Regulation of cell adhesion
In tumor progression, microenviromental factors such as cell adherence to extracellular matrix, host–tumor interactions, degradation of matrix components, migration and invasion are essential for acquisition of the metastatic phenotype. Changes in cadherins, gap junctions and MMP expression are major factors in ovarian carcinoma progression (Spinella et al. 2003). In epithelial cancer, acquisition of invasiveness is often accompanied by the loss of the epithelial features and the gain of a mesenchymal phenotype, a process known as epithelial to mesenchymal transition (EMT). This change is characterized by disassembling of GJIC, tight junctions (TJ) and adherent junctions (AJ), reorganization of cell substrate adhesion complexes, loss of cell polarity and significant remodelling of the cytoskeleton. These factors enable tumor cells to overcome microenvironmental control from the host, and to invade and metastatize (Roskelley & Bissell 2002). A primary event that governs EMT is the disruption of the E-cadherin-mediated stable interactions between the cells (Thiery 2002, Bissell & Radisky 2001, Conacci-Sorrell et al. 2002). Loss of E-cadherin can be accompanied by increased expression of mesenchymal N-cadherin that promotes inappropriate signals through interaction with the stromal cells (Cavallaro & Christofori 2004). Normal cells of the ovarian surface epithelium express little or no E-cadherin (Auersperg et al. 1999). Although many primary ovarian carcinomas express E-cadherin at the cell surface and in the cytoplasm, cell-surface expression of E-cadherin is reduced in many advanced carcinomas, confirming the paradigm of EMT as an integral component of the acquisition of the invasive phenotype (Roskelley & Bissell 2002, Faleiro-Rodrigues et al. 2004). Interestingly, a recent study demonstrated that immunoreactivity for E-cadherin and α-, β-, γ-catenin was significantly increased in the metastatic lesions compared with the respective primary ovarian tumors (Imai et al. 2004). In metastatic colonization, the reversed EMT process promotes establishment of secondary carcinomas. It therefore makes sense that the initial invasion stage is likely to require a rapid and significant repression of E-cadherin, while the regrowth of the secondary tumor as metastasis requires the re-expression and maintenance of E-cadherin. This notion is supported by the fact that E-cadherin downregulation in most carcinomas is a transient and dynamic event (Zhou & Hung 2005). In ovarian carcinoma cells, activation of the ETAR pathway by ET-1 contributes to disruption of normal host–tumor interactions by downregulating the expression of E-cadherin and associated β-catenin adhesion proteins. (Rosanò et al. 2005). ET-1 induced expression of the transcription factor Snail, which has been identified a potent repressor of E-cadherin expression (Battle et al. 2000, Cano et al. 2000), closely correlates with downregulation of β-cadherin. ET-1 also causes a concomitant upregulation of the mesenchymal N-cadherin, which can mediate homo-typic adhesive interactions as well as heterotypic cell–cell interactions (Savagner 2001). These effects are associated with ETAR-mediated enhancement of cell migration and invasiveness (Rosanò et al. unpublished results).
In this context, ET-1 through ETAR regulates the interactions between tumor cells and the surrounding normal microenvironment by regulating changes in cell surface adhesion and communication molecules and tumor proteinases (Fig. 2⇓). These data have been supported by analysis of a genome wide expression profile of advanced stage serous ovarian cancer (Donninger et al. 2004). Microarray results and bioinformatic analysis have identified ET-1 as a key gene that activates signaling pathways controlling ovarian cancer migration, spread and invasion.
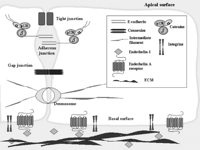
Mechanism of cell–cell and cell–ECM interactions regulated by ET-1 controlling epithelial cell plasticity. Binding of ET-1 to the ETAR can induce activation of tumor-associated proteinases that degrade the extracellular matrix (ECM). ET-1 also regulates adherens junctions, consisting of extracellular E-cadherin dimers that are anchored through catenins to actin filaments and constitute physical junctions, as well as gap junctions, that interconnect the cytoplasms of adjacent cells and are formed by the association of two hemichannels, each a hexamer of connexin subunits.
Targeting endothelin receptor as novel approach in ovarian carcinoma treatment
In view of its contributions to ovarian cancer progression by inducing cell proliferation, survival, angiogenesis and invasiveness, the ETAR has been proposed as a novel target for anticancer therapy. The recent identification of highly-selective small molecules that inhibit ligand-induced activation of ETAR offers the possibility of testing this therapeutic approach in a clinical setting (Remuzzi et al. 2002). Among the ETAR antagonists, ZD4054 is an orally active ETAR antagonist in early clinical development for the treatment of cancer (Morris et al. 2005). ABT-627 (Atrasentan) is an orally bioavailable endothelin antagonist that potently and selectively binds to the ETAR, blocking signal transduction pathways implicated in cancer, cell proliferation and other host-dependent processes promoting cancer growth (Carducci et al. 2002, 2003).
In ovarian cancer, ABT-627 inhibits in vitro cell proliferation, the ET-1 mediated protection against paclitaxel-induced apoptosis and the release of VEGF. A co-operative proapoptotic and VEGF inhibitory effect was observed when ABT-627 was used together with paclitaxel. Treatment with ABT-627 produced a 65% tumor growth inhibition in well established HEY xenografts. This treatment, which was generally well tolerated with no detectable signs of acute or delayed toxicity, was long-lasting and comparable to that achieved by paclitaxel. Immunohistochemical analysis of the xenografts revealed a marked reduction in the levels of COX-2, VEGF and MMP-2 in the treated mice. Tumor-induced vascularization, quantified as MVD, was directly proportional to the expression of VEGF. In addition the significant increase in the percentage of TUNEL-positive cells was found (Rosanò et al. 2003b). The tumor growth inhibition induced by ABT-627 was also associated with a reduction of Cx43 phosphorylation, and N-cadherin expression, and with an increase Cx43-based inter-cellular communication and E-cadherin and β-catenin expression. These findings suggest that ETAR blockade also contributes to the control of ovarian carcinoma growth and progression by preventing the loss of GJIC and AJ (Spinella et al. 2003, Rosanò et al. 2005). More marked and prolonged tumor growth inhibition (90% of controls) was obtained by combined treatment of ABT-627 with paclitaxel, with no toxicity and 40% complete tumor regressions. Almost complete inhibition of VEGF, MMP-2 expression, and tumor neovascularization, and an increase in apoptosis, were observed following combined treatment with ABT-627 and paclitaxel. The co-operative antitumor effect of combination therapy in which ETAR antagonist, by increasing the commitment of tumor cells towards apoptosis, potentiates the therapeutic efficacy of conventional cytotoxic drugs, offers a rationale for its clinical evaluation in malignancies expressing the ETAR (Bagnato et al. 2002, Del Bufalo et al. 2002, Rosanò et al. 2003b). These findings demonstrate the antitumor, anti-angiogenic and apoptotic activities of ABT-627 in vivo. This provides a rationale for the clinical evaluation of this molecule, alone and in combination with other therapies, in patients with ovarian tumors and potentially in other epithelial tumors that overexpress functional ETAR.
Metastasis is thought to be a multistep process requiring the concerted actions of several genes. EMT and metastasis are finely tuned processes that involve a complex signaling network causing cell fate changes and key alterations in cell behaviour. These findings show that ETAR-driven EMT requires activation of multiple pathways that in turn control transcriptional programs mediating an invasive metastatic tumor phenotype. In conclusion, the multiple molecular pathways elicited by ET-1 are triggered by the ETAR, leading to activation of the known molecular effectors involved in ovarian cancer progression, including tumor proteases, cell–cell adhesion and communication molecules. Blockade of the ETAR by small molecules results into inhibition of ovarian carcinoma growth and progression in vitro and in vivo, thus offering an unprecedented opportunity for targeted therapy in the treatment of this malignancy.
The role of the ET-1 system and the therapeutic relevance of ET-1 receptor antagonists in a range of malignancies requires future investigation that may lead to a new generation of molecular targeted therapies for cancer (Table 1⇓) (Nelson et al. 1995, 1996, 1997, Guise et al. 2000, Venuti et al. 2000, Bagnato et al. 2001, 2002b, 2003, Rosanò et al. 2003a, Wulfing et al. 2003, 2004, 2005).
Role of ET-1 axis in different malignancies
Acknowledgments
We thank V Di Castro and M R Nicotra for their contributions to in vitro and in vivo studies, P G Natali for critical reading and insightful comments, M V Sarcone for secretarial assistance and Abbott Laboratories, (Abbott Park, IL, USA) for kindly providing atrasentan.
Funding
This work was supported by the Associazione Italiana Ricerca sul Cancro, Ministero della Salute and CNR-MIUR. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
- © 2005 Society for Endocrinology










