- Accepted Preprint first posted online on 6 January 2011
Thyroid hormones and fetal neurological development
- 1School of Medicine, The University of Queensland, Herston, 4006 Brisbane, Queensland, Australia
2Conjoint Endocrine Laboratory, Queensland Institute of Medical Research, Bancroft Centre, Royal Brisbane and Women's Hospital, 300 Herston Road, Herston, 4029 Brisbane, Queensland, Australia
3Disciplines of Medicine, Obstetrics and Gynaecology, The University of Queensland, Herston, 4006 Brisbane, Queensland, Australia
- (Correspondence should be addressed to J Patel at Conjoint Endocrine Laboratory, Queensland Institute of Medical Research, Bancroft Centre, Royal Brisbane and Women's Hospital; Email: jatin.patel{at}qimr.edu.au)
Abstract
The development of fetal thyroid function is dependent on the embryogenesis, differentiation, and maturation of the thyroid gland. This is coupled with evolution of the hypothalamic–pituitary–thyroid axis and thyroid hormone metabolism, resulting in the regulation of thyroid hormone action, production, and secretion. Throughout gestation there is a steady supply of maternal thyroxine (T4) which has been observed in embryonic circulation as early as 4 weeks post-implantation. This is essential for normal early fetal neurogenesis. Triiodothyronine concentrations remain very low during gestation due to metabolism via placental and fetal deiodinase type 3. T4 concentrations are highly regulated to maintain low concentrations, essential for protecting the fetus and reaching key neurological sites such as the cerebral cortex at specific developmental stages. There are many known cell membrane thyroid hormone transporters in fetal brain that play an essential role in regulating thyroid hormone concentrations in key structures. They also provide the route for intracellular thyroid hormone interaction with associated thyroid hormone receptors, which activate their action. There is a growing body of experimental evidence from rats and humans to suggest that even mild maternal hypothyroxinemia may lead to abnormalities in fetal neurological development. Our review will focus on the ontogeny of thyroid hormone in fetal development, with a focus on cell membrane transporters and TR action in the brain.
Introduction
While the impaired mental and physical development of inhabitants of the iodine-deficient Alps was recognized as cretinism hundreds of years ago, the link between iodine deficiency, hypothyroidism, and impaired neurologic development was made very much later ( Cranefield 1962, Morreale de Escobar et al. 2004). Thyroid hormones (thyroxine (T4) and triiodothyronine (T3)) are essential for the development and maintenance of normal physiological processes, especially those of the central nervous system, where thyroid hormones assist in brain maturation throughout gestation ( Joffe & Sokolov 1994, Neale et al. 2007). Thyroid hormones primarily regulate genes involved in myelination and neuronal\glial cell differentiation ( Bernal 2005). Delivery of thyroid hormones to the fetal brain is a complex process requiring, at different times, expression of brain thyroid hormone receptors (TRs), materno-fetal thyroid hormone and iodide transport, an intricate system of endocrine feedback (the hypothalamic–pituitary–thyroid (HPT) axis and thyroid hormone metabolism by liver and brain deiodinase enzymes (deiodinase type 2 (D2) and deiodinase type 3 (D3)) to ensure basal levels are sustained ( Zoeller et al. 2007)).
The fetal thyroid gland reaches maturity by week 11–12, close to the end of the first trimester and begins to secrete thyroid hormones by about week 16 ( Obregon et al. 2007). During this period, an adequate supply of maternal thyroid hormones must be sustained to ensure normal neurological development. Hypothyroid fetuses suffer various postnatal disorders including mental retardation, deafness, and spasticity. Severe iodine deficiency, which causes both maternal and fetal hypothyroidism, is, worldwide, the most common cause of mental retardation ( Glinoer 2001, Morreale de Escobar et al. 2004, Pearce 2009). If thyroid hormone replacement for congenital hypothyroid babies is not initiated soon after birth, further impairment of cognitive development occurs. More recent evidence suggests that even mild reductions in maternal thyroid hormone levels in early pregnancy are associated with reduced IQ in offspring ( LaFranchi & Austin 2007, Gyamfi et al. 2009). The molecular mechanisms by which thyroid hormones affect fetal neurological structures are still not well understood. This review will briefly discuss some current aspects of the role of thyroid hormones in fetal neurological development, focusing on TH transporters and receptor activation in the brain.
Ontogenesis of thyroid hormone action in fetal development
There is growing evidence that thyroid hormones act on embryological and fetal tissues early in development. Thyroid hormone and associated receptors are already found in human fetal tissues prior to the production and secretion of fetal thyroid hormones at 16–18 weeks of gestation, as evidenced by detection of T4 and T3 in the human cerebral cortex by week 12 gestation ( Calvo et al. 2002, Kester et al. 2004). This is confirmation that active transport of maternal thyroid hormone across the placenta is occurring during this crucial period of gestation and highlights the need for maternal thyroid hormones to be at optimal levels at that time ( Fig. 1; Bernal 2007). Following onset of active T4 secretion by the fetus, levels of T4 in fetal tissues parallel those in fetal plasma. T4 levels are, however, low, reflecting active type 3 deiodination in the fetus ( Ruiz de Ona et al. 1988, Obregon et al. 2007). D3 converts T4 to the biologically inactive reverse T3. In contrast to tissue T4 levels, T3 concentrations vary in different tissues, for example levels are low in fetal liver and plasma and high in brain and brown adipose tissue ( Obregon et al. 2007). These differences have been attributed to variations in the activity of the D2, which converts T4 to biologically active T3. This suggests an important role for T3 in brain developmental and maturation processes. D3 is also active in placenta, ensuring the fetus is not exposed to excessive amounts of maternal T4 ( Galton 2005).
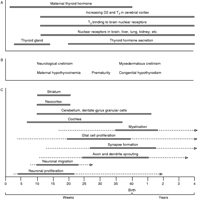
(A) The ontogeny of fetal thyroid function and expression of thyroid hormone receptors and deiodinase enzymes during gestation and early years postnatal are demonstrated. (B) Critical time points during gestation that require thyroid hormone action for fetal neurological development. Neurological abnormalities can be seen if maternal or fetal hypothyroidism is present during gestation. (C) Rodent studies, where time-specific actions of thyroid hormones on precise neurological and auditory structures were observed. Adapted image from Bernal (2007).
Much of the information known about thyroid hormones and brain development has been derived from rodent experiments. As in humans, normal neurological development depends on thyroid hormone. TRs and deiodinase enzymes are expressed in the early brain before the thyroid gland develops ( Obregon et al. 2007). The critical time period for thyroid hormone action in rat brain is estimated to extend from around embryonic day 18 (E18) to postnatal day 21–25 (P21–25; Porterfield & Hendrich 1993). Abnormalities in brain development in hypothyroid rats are mostly seen in the postnatal period and are demonstrated by reduced maturation of key structures such as the cerebellum, where delayed granular cell migration and Purkinje cell maturation are prevented ( Koibuchi et al. 2003). Although this may suggest that rat brain is more affected by thyroid hormone action after birth, it should be recognized that TRs are seen very early in rat development (discussed below). Rat studies also indicate that the developing brain is dependent on a supply of T4, which is locally deiodinated to T3, and that replacement with T3 does not adequately replenish brain T3 levels ( Calvo et al. 1990). This emphasizes the need for maternal T4 levels to be maintained to ensure normal fetal brain development. This also explains why even minimally reduced maternal T4 levels in early pregnancy can result in adverse outcomes to the offspring ( Lavado-Autric et al. 2003, Auso et al. 2004).
Deiodination of thyroid hormones
As mentioned above, locally generated T3 in the brain from maternally transported T4 has been reported to be essential for normal early brain development ( Zoeller 2010). Almost 80% of brain T3 is produced locally by D2 ( Crantz et al. 1982). D2 is found almost exclusively in astrocytes, whereas TRs are highly expressed in oligodendrocytes and neurons ( Guadano-Ferraz et al. 1997). In the fetal rat, D2 expression is first seen at E16.5 and increases steadily until P15. In human brain, D2 expression is first detectable in the cerebral cortex in the first trimester of pregnancy at the same time that T3 can be measured there ( Chan et al. 2002). D2 is largely responsible for maintaining appropriate concentrations of T3 during fetal brain development ( Guadano-Ferraz et al. 1999). D2 knockout (KO) mice also demonstrate impaired cochlea and visual development, poor thermal regulation, reduced anxiety, and crucially pituitary resistance to T4, further highlighting the importance of D2 in developmental processes ( Obregon et al. 1991). In cases of hypothyroidism, D2 expression and activity are up-regulated, enhancing T3 supply, whereas in hyperthyroidism, the opposite is true ( Burmeister et al. 1997). Interestingly, however, a more recent study in D2KO mice demonstrated that locomotor activity and learning and memory skills were normal despite low local T3 generation ( Galton et al. 2007).
Raised brain levels of thyroid hormone in the fetus can also cause neurological damage. D3 plays an important role in fetal neurological development by ensuring that safe levels of thyroid hormones are maintained ( Gereben et al. 2008). D3 is highly expressed in neurons and in contrast to D2 is positively regulated by thyroid hormone. D3KO mice demonstrate significant increases in perinatal mortality and an abnormal HPT axis ( Hernandez et al. 2006, 2007). Furthermore, D3KO mice are born hypothyroid with impaired growth and fertility ( Galton 2005). They exhibit excessive T3-responsive gene activation during development and reduced activation later in life ( Horn & Heuer 2010).
Thyroid hormone transporters in the brain
Over the last decade, the notion of thyroid hormone uptake into cells by passive diffusion has been dismissed following the discovery of a number of thyroid hormone cell membrane transporters. Thyroid hormones are amino acid derivatives, and several classes of transmembrane transporter proteins can transport thyroid hormones, including organic anion transporters 2 and 3 (Oatp2 and Oatp3), L-type amino acid transporters (Lat1 and Lat2), and monocarboxylate transporters 8 and 10 (MCT8 and MCT10) ( Abe et al. 1998, Friesema et al. 2001, 2003, 2008). For thyroid hormone to gain access to the brain during maturation, it must pass through many different cell types which express the various cell membrane transporters. For rodents, the preferred route of TH entry is through the cerebral circulation and its blood–brain barrier, with some thyroid hormone entering the cerebrospinal fluid (CSF) via the choroid plexus ( Dratman et al. 1991, Horn & Heuer 2010). The T4-binding protein, transthyretin, produced in significant amounts by the choroid plexus, has been implicated in this transport, and transthyretin may be involved in delivery of CSF thyroid hormone to the brain ( Patel et al. 2010). T4 transported to the brain via blood circulation passes through endothelial cells and is taken up by astrocytes, probably through the Oatp1c1 cell membrane transporter ( Fig. 2; Hernandez et al. 2007). Within the astrocyte, T4 is deionated via D2 to produce T3, which then exits the cell possibly via MCT8 (yet to be elucidated) and is taken up via MCT8 by oligodendrocytes and neurons ( Fig. 2; Heuer et al. 2005).
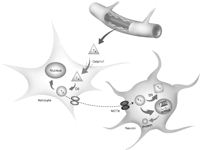
T4 is transported to the brain thyroid hormone-binding proteins such as transthyretin (TTR), where T4 then passes out through endothelial cells lining the blood vessels. T4 is rapidly transported through the cell membrane transporter, Oatp1c1, located on the surface of astrocytes. T4 is then metabolized intracellularly by D2 to T3, where it can then be transported out from astrocytes by an as yet unidentified cell membrane transporter. Within the brain parenchyma, T3 is then promptly uptaked by neurons and oligodendrocytes via the MCT8 cell membrane transporter. Within the cell, T3 can either translocate and bind to thyroid hormone receptors (TRs), resulting in thyroid hormone action, or be metabolized via D3 to biologically inactive T2.
In humans, the lack of MCT8 leads to greater neurological damage than in rodents, suggesting that other but as yet undefined transporters are present in rodents. Lat2 has been proposed as a candidate, which although expressed in developing rat brain is not present in human developing neurons ( Wirth et al. 2009). In humans, the absence of the MCT8 transporter (the Allan–Herndon–Dudley syndrome) results in X-linked moderate to severe mental retardation and muscle hypotonia and hypoplasia ( Dumitrescu et al. 2004, Friesema et al. 2004, Schwartz et al. 2005). Raised serum T3 concentrations and low T4 levels are present, but TSH levels are normal suggesting a role for MCT8 in pituitary thyroid hormone uptake ( Friesema et al. 2006). Patients also display altered and delayed maturation of myelination ( Namba et al. 2008). More than two dozen MCT8 gene mutations have been reported linked to the X-chromosome ( Friesema et al. 2010). Almost all are missense mutations resulting in reduced MCT8 expression, cell surface translocation, and specific substrate transport deficits, consequently producing a complete loss-of-function phenotype in patients ( Kinne et al. 2009). MCT8 may transport other molecules essential for brain maturation and which could explain the difference between human and rodent brain maturation in the presence of MCT8 transporter mutations.
TR genes and activity
There are two TR genes (THRA and THRB), which encode for the four isotypes of TR (TRα1, TRβ1, TRβ2, and TRβ3) ( Bernal 2007). The four isotypes all bind to T3 and DNA and drive intracellular thyroid hormone action. Several isoforms of the TR from alternate RNA splicing are also seen. Two of these are the TRα2 (c-erbAα2) and truncated TRβ3 (ΔTRβ3), which are known as nonreceptor proteins ( Bassett et al. 2003). TRα2 has a conserved DNA-binding region but is unable to bind T3. Conversely, ΔTRβ3 is able to bind T3 but not to DNA. The physiological role of these nonreceptor proteins is still unknown ( Forrest & Vennstrom 2000, O'Shea & Williams 2002, Bernal 2007).
TRs mediate their actions following homodimerization or heterodimerization with retinoic acid receptors (RXR), which then bind to specific sequences known as thyroid response elements (TREs) in the regulatory regions of target genes ( Wagner et al. 1995, Feng et al. 1998). A multitude of transcription factors are involved. Without the presence of T3, the unliganded receptor (aporeceptor) recruits corepressors, such as nuclear receptor corepressors (NCoR) or silencing mediator for retinoic and thyroid receptor (SMRT) and histone deacetylases, which retain the chromatin in a compact repressed position ( Bernal 2007). However, in the presence of T3, the binding of the hormone to TR initiates transcription by the release of corepressors and recruiting coactivators (steroid receptor coactivators 1, SRC-1), histone acetylases (CREB-binding protein, CBP; p300; and mammalian homolog of the yeast transcriptional activator GCN5, pCAF), and other mediators, all of which assist in the access of transcription apparatus to the promoter regions ( Fig. 3; Bernal 2007, Cheng et al. 2010).
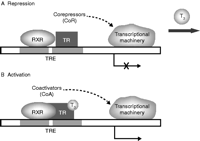
Schematic representation of thyroid hormone receptor (TR) action. (A) Without the presence of the T3 ligand, corepressors (CoR) (nuclear receptor corepressors (NCoR) or silencing mediator for retinoic and thyroid receptor (SMRT)) and histone deacetylases are recruited by TR to prevent transcription and translation of target genes is demonstrated. (B) A bound TR with T3 results in the recruitment of coactivators (CoA) (steroid receptor coactivators 1, SRC-1) and histone acetylases (CREB-binding protein, CBP; p300; and mammalian homolog of the yeast transcriptional activator GCN5, pCAF) as well as the release of CoR, resulting in changes in expression of target genes is demonstrated.
Both TR genes, THRA and THRB, are found within the brain. In adult rat brain, more than 70–80% of all TRs present are in the TRα1 isoform. The receptor proteins are predominantly found in the cerebrum and cerebellum ( Ercan-Fang et al. 1996). It has been postulated that the TRα1 isoform also plays an important role during fetal brain development, both in humans and in rodents. This is evidenced by detection in fetal rat brain of mRNA E11.5 in the neural tube and E12.5 in the diencephalon and ventral rhombencephalon ( Bradley et al. 1992). It is believed that the TRs found in these regions mediate the biological effects of T3 that has been locally generated from transported maternal T4, early in gestation. Conversely, TRβ isoforms are expressed more postnatally within specific neuronal populations such as hippocampal pyramidal and granule cells, paraventricular hypothalamic neurons, and cerebellar Purkinje cells ( Bradley et al. 1989, Horn & Heuer 2010). Studies in TRβ KO mice have shown that these isoforms are predominant in mediating thyroid hormone effects on the development of the vision and auditory systems ( Jones et al. 2003).
There is a growing body of evidence suggesting that deficiencies in TR may not lead to abnormal brain development as seen in hypothyroidism, which suggests that the lack of the ligand (T3) is more detrimental than being TR deficient ( Bernal 2007). This has been demonstrated by Bernal et al. in TRα1 KO mice, which have normal cerebellar development, even though the mice were hypothyroid ( Morte et al. 2002). Another interesting study has demonstrated that TRα1 deletion did not affect cerebellar granule cell migration during development, which is now known to take place in the absence of both receptor and T3 ( Yacubova & Komuro 2002). It has been postulated by Bernal et al. that this result could well demonstrate that in vivo thyroid hormones are permissive when the TR are present, releasing any blocking function that TR may be regulating ( Bernal 2007). In contrast, the absence of ligand could lead to aberrant receptor signaling, which has been demonstrated with hypothyroid wild-type animals with cell migration defects. This phenomenon has also been demonstrated using pheochromocytoma PC12 cells. When exposed to nerve growth factors, these cells differentiate into neurons. Specifically, T3 has no effect on this process; however, exogenous unliganded TRα1 will block this process unless T3 is added to the culture medium ( Munoz et al. 1993). This has led to the hypothesis that the aporeceptor has some transcriptional activity that may play an as yet unrecognized role in developmental processes. This also poses the possibility of the aporeceptor possibly repressing specific gene expression until T3 becomes available to the cells in a time-specific manner.
Further analysis into the role of TR and brain development has been studied using mouse mutants expressing dominant negative TR, where DNA binding is conserved with reduced or absent T3-binding capacity. In two different models of TRβ mutant mice, impaired cerebellar Purkinje cell development and maturation and motor deficits were observed, with cerebellar morphology similar to that seen in hypothyroidism ( Forrest & Vennstrom 2000). This also confirms the dominant role of TRβ isoforms within the Purkinje cells of the brain. In TRα mutant models where T3-binding affinity was reduced by almost tenfold, mice showed significant growth retardation and cardiac abnormalities, with the heterozygous strain also demonstrating similar cerebellar abnormalities seen in hypothyroidism ( Tinnikov et al. 2002). These mice also displayed striking anxiety-related behavior as evidenced by time spent motionless and reduced exploratory behavior compared to the wild-type controls ( Tinnikov et al. 2002). This evidence further strengthens the hypothesis that TR play roles in fetal brain development and maturation, especially within the hippocampus, and in maintaining adult brain function ( Bernal 2007). Further insights into phenotypic differences between these mutant mice strains are of particular interest, with additional work now being conducted in generating cell-specific TR mutant models ( Quignodon et al. 2007, Flamant & Quignodon 2010).
TRs have also been observed within the cytosol, where they interact with p85, a regulatory subunit of phosphatidylinositol 3-kinase ( Moeller et al. 2006). This affects the AKT pathway, resulting in induction of nitric oxide synthesis in endothelial cells in response to middle cerebral artery occlusion ( Hiroi et al. 2006). This highlights the need for further investigation into the complex roles that TR play in physiological and pathological processes.
Conclusion
Our understanding of the complex processes involved in ensuring normal fetal development is increasing. Knowledge of the key relationships between thyroid hormone and brain development has progressed significantly over the past decade. However, many issues regarding TR and thyroid hormone cell membrane transporters have yet to be clarified. Differences between rodent and human development are significant and need to be further explored to ensure that the relationships between thyroid hormone and human brain development are better understood.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
- Received in final form 21 December 2010
- Accepted 6 January 2011
- © 2011 Society for Endocrinology









