- Made available online as an Accepted Preprint 12 April 2010
- Accepted Preprint first posted online on 12 April 2010
Generating pancreatic β-cells from embryonic stem cells by manipulating signaling pathways
- Diabetes Research Group, Division of Reproduction and Endocrinology, King's College London, Hodgkin Building (2.10N), Guy's Campus, London SE1 1UL, UK
- (Correspondence should be addressed to S Champeris Tsaniras; Email: spyridon.champeris-tsaniras{at}kcl.ac.uk)
Abstract
Type 1 diabetes results from an insufficiency of insulin production as a result of autoimmune destruction of the insulin-secreting pancreatic β-cells. It can be treated by transplantation of islets of Langerhans from human donors, but widespread application of this therapy is restricted by the scarcity of donor tissue. Generation of functional β-cells from embryonic stem (ES) cells in vitro could provide a source of an alternative graft material. Several ES cell differentiation protocols have reported the production of insulin-producing cells by mimicking the in vivo developmental stages of pancreatic organogenesis in which cells are transitioned through mesendoderm, definitive endoderm, foregut endoderm, pancreatic endoderm, and the endocrine precursor stage, until mature β-cells are obtained. These studies provide proof of concept that recapitulating pancreatic development in vitro offers a useful strategy for generating β-cells, but current differentiation protocols employ a bewildering variety of growth factors, mitogens, and pharmacological agents. In this review, we will attempt to clarify the functions of these agents in in vitro differentiation strategies by focusing on the intracellular signaling pathways through which they operate – phosphatidylinositol 3-kinase, transforming growth factor β, Wnt/β-catenin, Hedgehog, and Notch.
Introduction
Type 1 (insulin-dependent) diabetes mellitus is a T cell-mediated autoimmune disease resulting in the destruction of insulin-secreting β-cells which are found exclusively in the pancreatic islets of Langerhans (Eiselein et al. 2004). It is a complex disorder whose pathogenesis involves B and T lymphocytes, macrophages, dendritic cells, and β-cell autoantigens, as well as multiple environmental and genetic components (Mandrup-Poulsen 2003, Yoon & Jun 2005). β-Cell destruction is thought to be induced by cytokines produced by activated β-cell-specific autoreactive T-cells (Eiselein et al. 2004). The ability of the endocrine pancreas to produce insulin decreases until insulin production is no longer sufficient to maintain whole-body glucose homeostasis, at which point the patient presents with the clinical symptoms of type 1 diabetes. In brief, reduced circulating insulin causes decreased cellular uptake of glucose by peripheral tissues as well as hepatic overproduction of glucose, both of which result in hyperglycemia and the associated glycosuria, polyuria, and polydipsia. In the UK, type 1 diabetes affects ∼15–20 per 100 000 individuals a year with a relative increase in incidence around 3% yearly. Worldwide, the increase in incidence ranges around the same levels, indicating that it will be 40% higher in 2010 compared to 1998 (Onkamo et al. 1999).
A substantial amount of research has focused on providing a permanent cure for diabetes by replacing the lost β-cells. At present, the most promising new therapies are based on replenishing the β-cell mass. The development of the Edmonton protocol for pancreatic islet transplantation proved that cadaveric human islets can be grafted to diabetic patients using a minimal, glucocorticoid-free immunosuppressive regimen and provide insulin independence, if only for a limited period of time (Shapiro et al. 2000, Ryan et al. 2005). However, the shortage of cadaver pancreata for islet isolation has resulted in the search for alternative sources of insulin-producing cells.
A variety of pluripotent cell types have been suggested as potential starting material from which to generate an unlimited supply of insulin-secreting cells (see Seaberg et al. 2004, Suzuki et al. 2004, Hori et al. 2005), but this review will focus on the progress that has been made using embryonic stem (ES) cells (Evans & Kaufman 1981, Thomson et al. 1998). This area of research was initiated around a decade ago by proof-of-concept studies demonstrating the generation of insulin-expressing cells from mouse ES (mES) cells (Soria et al. 2000, Lumelsky et al. 2001). Lumelsky et al. (2001) described the first culture-based protocol for the in vitro generation of pancreatic cells from mESCs using a five-step protocol that was reported to generate insulin-expressing cells by selecting for nestin-positive progenitor cells. The reliability of this strategy was subsequently questioned when it was suggested that the insulin expression reflected uptake of insulin from the culture medium rather than de novo synthesis associated with insulin gene expression (Rajagopal et al. 2003, Hansson et al. 2004, Sipione et al. 2004, Paek et al. 2005). Despite this, several other groups modified the initial Lumelsky protocol and reported improved β-cell differentiation, although these protocols should be treated with some caution. For example, Hori et al. (2002) treated mESCs with an inhibitor of phosphatidylinositol 3-kinase (PI3K) and reported improved results over the original Lumelsky protocol. In a similar approach, Ku et al. (2004) increased the number of insulin-positive cells by the inclusion of activin βB, exendin-4, and nicotinamide to the culture medium.
Other groups tried to direct the differentiation of the mES cells by overexpressing transcription factors critical for β-cell development, such as Pdx1 (Blyszczuk et al. 2003, Miyazaki et al. 2004), Pax4 (Blyszczuk et al. 2003), or Nkx2-2 (Shiroi et al. 2005), and reported the generation of insulin-expressing cells. This approach seemed to be the most promising strategy until recently, when safety concerns over therapeutic applications of genetically modified cells (Strulovici et al. 2007) led to a shift towards inducing differentiation solely by exposure to extracellular factors. D'Amour et al. at Novocell developed an in vitro differentiation protocol (Novocell protocol) using human ES cells (hESCs) that mimicked the in vivo developmental stages of pancreatic organogenesis. Thus, the aim was to provide sufficient cues to enable cells to transition through mesendoderm, definitive endoderm (DE), gut tube endoderm, pancreatic endoderm, and endocrine precursor stages, resulting in insulin-expressing cells with a reported insulin content approaching that of adult islets (D'Amour et al. 2006). Since then, several other groups devised similar differentiation protocols addressing the main developmental stages of the endocrine pancreas, especially definitive and pancreatic endoderm (Schroeder et al. 2006, Blyszczuk & Wobus 2007, Jiang et al. 2007a,b). More recently, the Novocell group has applied their protocol to generate endocrine precursors in vitro, and then grafted these progenitors into diabetic mice to promote their full maturation (Kroon et al. 2008), generating cells that secreted insulin at levels comparable to mature β-cells.
These proof-of-concept studies demonstrate that recapitulating in vitro the signals controlling the development of the endocrine pancreas in vivo offers a promising strategy for β-cell generation. To date, published β-cell differentiation protocols have used a bewildering variety of growth factors, mitogens, and pharmacological agents that act by exerting transcriptional control on the developmental process. Table 1 lists some of these differentiation agents, their cellular targets and, where known, their site of action in the differentiation process. In this review, we will attempt to clarify the use of these media supplements by focusing on the specific signaling pathways through which they operate, predominantly the PI3K, transforming growth factor β (TGFβ), Wnt/β-catenin, Hedgehog, and Notch pathways. Understanding the roles of these pathways in the differentiation of ES cells to functional β-cells will enable the future development of more precisely defined and efficient in vitro differentiation protocols.
Biologically active factors that have been reported to influence pancreatic endocrine differentiation in established embryonic stem (ES) cell differentiation protocols, with the reported mode of action (if known)
Modulators of signaling pathways
The PI3K signaling pathway
PI3Ks are a group of lipid kinases that have been implicated in numerous cellular processes controlling proliferation, apoptosis, DNA synthesis, cytoskeletal rearrangements, and cell migration. They are defined in three classes, based on their catalytic subunit. Class I are heterodimers, incorporating a catalytic as well as a regulatory subunit. Class II have a distinct C-terminal C2 domain, whereas Class III are the homologs of the yeast Vps34p protein. Once activated, PI3K generates the second messenger molecules phosphatidylinositol 3-phosphate, phosphatidylinositol 3,4-biphosphate, and phosphatidylinositol (3,4,5)-triphosphate. These molecules, in turn, transmit the signals downstream through various mediators, such as AKT and glycogen synthase kinase-3β (GSK-3β; reviewed by Vanhaesebroeck et al. (2001)).
PI3K signaling and the generation of β-cells
The PI3K signaling pathway has been shown to negatively regulate cellular differentiation in human and murine ES cells. Initially, this pathway was shown to be important in maintaining self-renewal of mESC cells through LIF-activated signaling (Paling et al. 2004). Consistent with this, it was later shown that activation of Akt, a major downstream component of this pathway, had similar effects in maintaining the pluripotency of murine and primate ES cells (Watanabe et al. 2006). More recently, McLean et al. (2007) have showed that suppression of PI3Ks facilitated the differentiation of hESCs into mesendoderm and DE under conditions in which activin–Nodal signaling was high. In an earlier study, suppressing the PI3K pathway was reported to promote endocrine differentiation of human fetal pancreatic cells (Ptasznik et al. 1997), perhaps suggesting an additional negative regulatory action of PI3K at a later stage in the β-cell differentiation pathway.
Modulators of the PI3K signaling pathway
The involvement of PI3K signaling in ES cell differentiation suggests that manipulation of this pathway may enhance ES cell differentiation towards a pancreatic lineage, with PI3K inhibitors being used at an early stage to specify DE and/or at a later stage to promote differentiation of endocrine precursors. Consistent with this, Hori et al. (2002) were the first to show that two direct pan inhibitors of the PI3K enzymes, LY294002 and wortmannin, enhanced endocrine differentiation of mESCs when used at the final stage of the differentiation protocol. More recently, Zhang et al. (2009) have used wortmannin to specify DE from hESCs, while in another study, an isoform-specific AKT inhibitor (AKTI-II), as well as LY294002, was able to drive differentiation of hESCs into DE (McLean et al. 2007).
A number of other PI3K pan inhibitors have not yet been used in pancreatic differentiation protocols, including ZSTK474, PX-866, and SF1126, nor have a number of isoform-specific PI3K inhibitors, including imidazopyridine-related compounds, morpholino-substituted pyridopyrimidine, quinolone and benzopyranone derivatives, quinazolinones and methylxanthines (Ward et al. 2003, Ito et al. 2007). The PI3K pathway can also be targeted at different sites. Compounds such as AKTI-1, AKTI-1,2, and staurosporine have been shown to inhibit the AKT mediator (Barnett et al. 2005). Other agents such as rapamycin and its derivatives can target MTOR, which is a downstream component of AKT (Tsang et al. 2007), although targeting downstream pathway components may be less effective than direct inhibition of PI3K.
The TGFβ signaling pathway
The TGFβ signaling pathway is complex, involving almost 30 different growth and differentiation factors. The signaling molecules belonging to the TGFβ superfamily have been subdivided into several subgroups, such as the bone morphogenetic protein, the Nodal, and the TGFβ subgroups. This pathway has been implicated in numerous cellular processes including proliferation, differentiation, apoptosis, cell migration, and adhesion (Massagué & Chen 2000). TGFβ signaling is mediated through two types of transmembrane serine/threonine kinase receptors, namely type I and type II. Binding of TGFβ family members to type II receptors results in phosphorylation and activation of type I receptors. Once activated, type I receptors phosphorylate specific intracellular mediators known as SMAD proteins, which act as signal transducers and translocate to the nucleus, where they regulate gene transcription (Massagué 1998).
TGFβ signaling and the generation of β-cells
The TGFβ pathway is central to the generation of mesendoderm and DE. Nodal ligands, which signal through the ActrIb (listed as Acvr1b in the MGI Database) and ALK7 type I receptors as well as the ActrIIa (Acvr2a) and ActRIIB type II receptors (Schier & Shen 2000), are thought to act as mesendoderm inducers. Thereafter, mesendoderm commitment to mesoderm or DE is thought to depend on the level of Nodal signaling, with high levels promoting an endodermal fate (Tremblay et al. 2000, Vincent et al. 2003). However, it is important to note that Nodal signaling can specify DE only when PI3K signaling is low, as described in section ‘PI3K signaling and the generation of β-cells’ (D'Amour et al. 2005, McLean et al. 2007). The Nodal pathway can also be activated by activin, which is known to signal through the same receptors (Chen et al. 2006). Consistent with this, several studies have shown that exogenous administration of activin A promotes DE differentiation of both mouse (Kubo et al. 2004, Tada et al. 2005, Yasunaga et al. 2005) and hESCs (McLean et al. 2007). As a result, this compound has been incorporated into several pancreatic differentiation protocols and, in conjunction with low PI3K signaling, has served as a first step to drive pluripotent cells towards DE formation (Shi et al. 2005, D'Amour et al. 2006, Jiang et al. 2007a,b, Phillips et al. 2007, Cho et al. 2008, Zhang et al. 2009).
Little is known about the possible roles of other TGFβ family members in promoting pancreatic differentiation. It was reported some time ago that TGFβ1 (TGFB1) enhanced the development of endocrine cells from mouse pancreatic fetal cells, with a more pronounced increase in β- and PP-cells (Sanvito et al. 1994), and this effect was subsequently reproduced by Tei et al. (2005). Another study showed that TGFβ2 (TGFB2) induced differentiation of mESCs into Pdx1-expressing endodermal cells (Shiraki et al. 2005), but several other growth factors from this family, including TGFα and TGFβ1, did not have any significant effect on promoting Pdx1 expression. In addition, the Pdx1-expressing cells induced by TGFβ2 expressed endodermal markers, but did not express insulin or glucagon, suggesting that TGFβ2 may be useful to promote an endodermal fate at an early stage in a differentiation protocol, whereas TGFβ1 may be useful after the specification of pancreatic or endocrine progenitors to enhance development of endocrine cells.
The importance of TGFβ signaling in defining DE was supported by a recent study in which two small molecules, named IDE1 and IDE2, were shown to direct differentiation towards the endodermal lineage with very high efficiency (Borowiak et al. 2009). Both molecules act by activation of the TGFβ signaling pathway, through SMAD2 phosphorylation, to drive ∼80% of mESCs and 60% of hESCs into DE, a higher efficiency than that achieved by activin A.
The Wnt/β-catenin signaling pathway
The Wnt/β-catenin pathway (Fig. 1) plays a key role in the development of many organs such as brain (McMahon & Bradley 1990, McMahon et al. 1992), kidney (Kispert et al. 1998, Itäranta et al. 2006), and pancreas (Wells et al. 2007). This pathway is activated when Wnt ligands bind to specific cell surface receptors, called frizzled (Fzd), leading to activation of the intracellular Ca2+, the planar cell polarity, or the β-catenin/canonical branch of the pathway. In the latter case, Fzd activation induces phosphorylation of intracellular proteins called dishevelled (Dvl), which, in turn, block the action of the GSK-3β enzyme which normally phosphorylates β-catenin. As a result of this, unphosphorylated β-catenin accumulates in the cytoplasm and is translocated into the nucleus, where it forms a complex with T-cell factor (Tcf)/lymphoid enhancer factor (Lef) proteins (such as TCF1, LEF, TCF3, and TCF4), as well as co-activators, to activate transcription of target genes (Peifer & Polakis 2000, Logan & Nusse 2004).
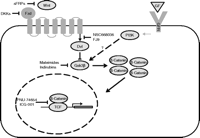
The Wnt signaling pathway. Wnt ligands bind to the frizzled receptor (Fzd) and activate the cytoplasmic protein dishevelled (Dvl) to inhibit the activity of glycogen synthase kinase-3β (GSK3β) or GSK3B. Gsk3β phosphorylates cytoplasmic β-catenin, targeting it for degradation at the proteosome. Inhibition of GSK3β activity therefore stabilizes the cytoplasmic pool of β-catenin, some of which then translocates to the nucleus to interact with members of the T-cell factor (TCF) family of transcription factors, leading to changes in the transcription of Wnt target genes. A variety of growth factors (GF), such as insulin, insulin-like growth factor 1, and epidermal growth factor, bind to specific receptors (GFR) and activate phosphatidylinositol 3-kinases (PI3K), which can interact with the Wnt signaling pathway at the level of GSK3β. However, it is unclear whether the same subcellular pool of GSK3β can be targetted by both PI3K and Wnt. The schematic also shows pharmacological agents that influence developmental processes through actions at specific stages of the Wnt signaling pathway (see main text for details).
GSK3β is also a component of the PI3K pathway, although it is unclear whether the same subcellular pool of GSK3β is targeted by both PI3K and Wnt. Several studies indicate that these two pathways crosstalk at the GSK3β level, suggesting that there is a single pool of the enzyme (Fukumoto et al. 2001, Almeida et al. 2005, He et al. 2007). However, a recent study has showed that GSK-3β could only be phosphorylated by PI3K when it is not bound to axin, whereas axin-bound GSK3β was dedicated to Wnt signaling (Ng et al. 2009).
The Wnt/β-catenin pathway and the generation of β-cells
Defining a precise role for Wnt signaling in the development of the endocrine pancreas is confounded by the complexity of the Wnt signaling pathway (Fig. 1), and the possibility of crosstalk between the canonical and non-canonical pathways. However, Wnt signaling has been implicated in both mesendoderm and foregut endoderm specification. Bakre et al. (2007) showed that activation of the pathway in mouse and hESCs causes mesendoderm-specific differentiation and generation of mesendodermal progenitor cells that can differentiate along various lineages. Similarly, Marikawa et al. (2008) reported that Wnt/β-catenin signaling was sufficient to induce the formation of mesendoderm in mouse embryonic carcinoma cells. In addition, a study by McLin et al. (2007) implicated Wnt signaling in foregut endoderm specification in Xenopus embryos. Thus, repressing Wnt signaling in the anterior endoderm was required for maintaining foregut fate, whereas high levels of Wnt signaling in the posterior endoderm enhanced intestinal development. They also postulated that Wnt factors from mesoderm can signal to the posterior endoderm to activate specific gene programs that inhibit foregut development, but cannot signal to the anterior endoderm, which is protected by secreting several Wnt antagonists. No similar studies have yet been carried out in other vertebrates, but several lines of evidence suggest that this is also the case in mice. For example, Wnt overexpression in the Pdx1 domain severely affects subsequent pancreatic development (Heller et al. 2002). In addition, deletion of Tcf1 (listed as Hnf1a in the MGI Database) and Tcf4 causes defects in hindgut development (Gregorieff et al. 2004), whereas expression of Tcf3 can be detected in the posterior but not the anterior DE (Merrill et al. 2004).
Modulators of the Wnt/β-catenin pathway
Given its role in mesendoderm and possibly in foregut endoderm specification, Wnt signaling should be activated at an early stage in an ES cell differentiation protocol and inhibited at a later stage, whereas a sustained activation of the pathway would be expected to enhance the formation of mesendoderm. This could be accomplished by using purified natural or recombinant Wnt-secreted proteins. So far, only three studies have employed Wnt agonism to generate mesendoderm from hESCs, and all of them have used Wnt3a (D'Amour et al. 2006, Cho et al. 2008, Kroon et al. 2008). However, more than 20 Wnt-secreted proteins have been identified, but their mechanisms of action are not well understood and their effects can be cell type and tissue specific (Naylor et al. 2000). Proteins which signal through β-catenin, such as WNT1, WNT2, WNT3, and WNT3a (van Gijn et al. 2002), would be predicted to be useful in endocrine pancreas differentiation protocols. Moreover, small-molecule Wnt agonists could also prove useful for this purpose. A large number of GSK3 inhibitors have been reported. Examples include maleimides such as SB-216763 (Coghlan et al. 2000) and bis-7-azaindolylmaleimide (Kuo et al. 2003), indirubins such as indirubin-3′-monoxime (Leclerc et al. 2001) and 6-bromoindirubin-3′-oxime (Meijer et al. 2003), pyrazolopyrimidine and benzimidazole–pyrazolopyrimidine derivatives (Peat et al. 2004a,b) as well as lithium ions (Stambolic et al. 1996), AR-A014418 (Bhat et al. 2003) and CHIR 98014 (Ring et al. 2003). However, these are not specific to GSK3β and can also inhibit GSK3α and other kinases, so more specific Wnt inhibition may be achievable by targeting other pathway components. For example, WAY-316606 specifically inhibits the Wnt antagonist sFRP-1 (Bodine et al. 2009), whereas QS11 synergizes with Wnt3a by binding to and inhibiting an ARFGAP protein, possibly resulting in increased β-catenin translocation (Zhang et al. 2007). Another compound, named compound 1, has been shown to induce Wnt signaling in a GSK3β-independent manner (Liu et al. 2005).
Although the available evidence also suggests an important role for Wnt suppression in the formation of posterior foregut endoderm, inhibition of the Wnt pathway has not yet been employed in in vitro differentiation protocols designed to generate endocrine pancreas. There is a long list of compounds that can inhibit this pathway at different levels. Some are endogenous secreted Wnt antagonists, including Dickkopfs (Dkks), Wnt inhibitory factor-1 (WIF-1), and secreted Fzd-related proteins (sFRPs). The most promising candidates include Dkk-1 and 4, which exert their actions by binding to a component of the Wnt receptor and preventing its activation, as well as sFRP2, sFRP3, and WIF-1, which bind to Wnt ligands (Kawano & Kypta 2003). Several natural and synthetic small-molecule inhibitors have also been identified, primarily because of their potential use as anti-cancer agents. For example, a high-throughput screening assay identified six natural compounds which prevented the formation of the Tcf/β-catenin complex, and the efficacy of these compounds was subsequently confirmed both in vitro and in vivo (Lepourcelet et al. 2004). Another Tcf/β-catenin complex inhibitor, PNU-74654, was identified by virtual screening of 17 700 synthetic compounds and shown to be active in vitro (Trosset et al. 2006). Another synthetic Wnt inhibitor, ICG-001, was reported to reduce Wnt signaling via binding to a transcriptional co-activator of β-catenin (Emami et al. 2004), while two other compounds, named NSC668036 and FJ9, were shown to disrupt the interaction between Fzd receptors and the Dvl PDZ domain (Shan et al. 2005, Fujii et al. 2007). The abundance of pharmacological modifiers of the Wnt signaling pathway (see Fig. 1) makes this an attractive target for inclusion in future in vitro differentiation protocols, as shown in Fig. 2.
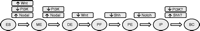
A proposed differentiation protocol for the generation of β-cells from embryonic stem cells. Manipulating the signaling cascades as shown is predicted to drive differentiation along the sequence of ES, embryonic stem cells; ME, mesendoderm; DE, definitive endoderm; PF, posterior foregut; PE, pancreatic endoderm; IP, islet precursors; BC, insulin-expressing β-cells. The role of PI3 and Shh (sonic hedgehog pathway) at the final stage of differentiation is not well understood (see main text for details).
The Hedgehog signaling pathway
The Hedgehog gene was identified almost 30 years ago and named after its ability to induce a hedgehog-like appearance in Drosophila (Nusslein-Volhard & Wieschaus 1980). Subsequent studies led to the identification of the Hedgehog homologs in other species, including mouse and human (Marigo et al. 1995). In vertebrates, three secreted hedgehog proteins have been identified – Desert, Indian, and Sonic Hedgehog (Echelard et al. 1993) – and it is now known that these proteins play vital roles in directing cell differentiation during embryonic development and organ formation. Hedgehog signaling is initiated when one of these ligands binds to specific cell surface receptors called Patched (Ptc). This leads to activation of Smoothened (Smo), a G-protein coupled-like receptor. Thereafter, signaling is mediated through a multiprotein complex, the Hedgehog signaling complex, which modifies the activity of Gli transcription factors in order to regulate expression of target genes (Villavicencio et al. 2000).
Hedgehog signaling and the generation of β-cells
The Hedgehog pathway has an important role to play in early pancreas development. Repression of Sonic hedgehog expression in the foregut region is essential for specification of the pancreatic domain. This repression occurs through activin βB and FGF2 signals, which are secreted by the notochord, thereby permitting expression of pancreatic transcription factors, such as Pdx1. Consistent with this, isolated notochord or purified activin βB and FGF2 have similar effects when applied to foregut endoderm cultures ex vivo (Hebrok et al. 1998). In accordance with this, increased Hedgehog activity during development impairs pancreas formation (Apelqvist et al. 1997, Kawahira et al. 2005), while inhibition of Hedgehog signaling causes an enlargement in islet and pancreas mass (Kim & Melton 1998, Hebrok et al. 2000). Hedgehog signaling may also be involved in the maintenance of pancreatic endocrine function. Thus, Thomas et al. (2000) used the INS-1 β-cell line to demonstrate that downregulation of hedgehog signaling decreased insulin secretion as a result of reduced insulin gene transcription. In a subsequent study, it was also shown that Pdx1, a key inducer of insulin expression, has Hedgehog-responsive regions within its promoter, and its expression is downregulated in the absence of Hedgehog. Conversely, ectopic Hedgehog expression was reported to increase both insulin and Pdx1 expression (Thomas et al. 2001).
Modulators of the Hedgehog signaling pathway
The developmental evidence suggests a dual role for Hedgehog signaling in a differentiation protocol for the generation of endocrine pancreas, with an initial inhibition of the pathway at an early stage of ES differentiation, followed by reactivation at a later stage.
The optimal timing for Hedgehog inhibition would be after DE has formed and Wnt signaling has been downregulated. In accordance with this, Shh is highly upregulated during embryoid body formation and DE induction, leading to repression of Pdx1 expression (Mfopou et al. 2005, 2007), implying that Hedgehog inhibitors would be beneficial at this stage. However, it is not immediately apparent which inhibitors would be most effective. Thus, when the Hedgehog inhibitor activin βB was applied during a mESC differentiation protocol at a late stage, it was reported to increase the percentage of insulin-expressing cells (Ku et al. 2004). In a later study, the same compound was shown to induce expression of the Pdx1 gene in cultured hESCs, but at the same time increased Shh expression (Frandsen et al. 2007). These unexpected results might indicate that the effects of activin βB are cell and tissue specific, whereas its differentiation-promoting activity might be Hedgehog-independent. In contrast, FGF2 (or bFGF) has been used extensively in differentiation protocols to expand pancreas progenitors and promote a pancreatic fate (Lumelsky et al. 2001, Miyazaki et al. 2004, Segev et al. 2004, Shi et al. 2005, Jiang et al. 2007a,b). It should be noted, however, that the effects of FGF2 seem to be concentration-dependent. In chicks, low concentrations of FGF2 increased Pdx1 and insulin expression and downregulated Shh expression, whereas high concentrations had the opposite effects (Hebrok et al. 1998). Other Hedgehog repressors that have been used in differentiation studies include Hedgehog-interacting protein (HIP) and cyclopamine. HIP is an endogenous hedgehog modulator which reduces Hedgehog signaling by direct binding to Hedgehog proteins (Chuang & McMahon 1999) and which has been reported to be efficient in increasing the percentage of insulin-positive cells in mESC cultures (Mfopou et al. 2007). Cyclopamine and its more potent derivative KAAD-cyclopamine are steroidal alkaloids known to block Smo, thereby preventing downstream signaling of the pathway (Taipale et al. 2000). Their efficiency has been demonstrated in both mouse (Skoudy et al. 2004, Serafimidis et al. 2008) and human pancreatic differentiation protocols (D'Amour et al. 2006, Cho et al. 2008). Several small-molecule inhibitors of the Hedgehog pathway could also prove useful in the derivation of β-cells from ES cells, including steroidal alkaloid inhibitors such as jervine, AY9944, triparanol, and U18666A (Cooper et al. 1998, Incardona et al. 2000). Chen et al. (2002) identified four naturally occurring small molecules (SANT-1 to 4) that were shown to directly inhibit Smo activity, as does a synthetic aminoproline named CUR61414 (Williams et al. 2003). Other agents, such as forskolin, 3-isobutyl-l-methylxanthine, and dibutyryl-cAMP (db-cAMP), act by increasing the activity of protein kinase A, which is known to antagonize Hedgehog signaling (Frank-Kamenetsky et al. 2002). Hedgehog downregulation was also shown to be promoted by JK184, an inhibitor of alcohol dehydrogenase 7 (ADH7), which is assumed to be correlated with Hedgehog signaling (Lee et al. 2007).
The reactivation of the Hedgehog pathway at a relatively late stage in the differentiation process may enhance insulin production after the endocrine progenitor stage. Hedgehog agonism can be achieved using purified recombinant SHH, IHH, or DHH proteins, all of which have been used in vitro (Dyer et al. 2001, Mfopou et al. 2007). Synthetic small-molecule agonists have also been identified, including SAG and Hh-Ag 1.1–1.5. SAG is a chlorobenzothiophene, which acts as an agonist at low concentrations but as an antagonist at higher concentrations (Chen et al. 2002). Hh-Ag 1.1 was identified by screening a library of 140 000 synthetic compounds and was reported to promote Hh signaling by stabilizing Smo. Subsequently, several other Hh-Ag 1.1 derivatives were synthesized. Hh-Ag 1.2 was shown to be the most stable, whereas Hh-Ag 1.3 showed the lowest toxicity in embryonic tissue cultures (Frank-Kamenetsky et al. 2002). In addition, purmorphamine, a 2,6,9-trisubstituted purine molecule, is a Hedgehog agonist which is thought to act by activating Smo or another upstream protein (Wu et al. 2004).
The Notch signaling pathway
The notch signaling pathway is a critical regulator of tissue development and cell fate decisions during embryogenesis. This pathway acts by either inhibiting or inducing the spread of cellular differentiation, known as lateral inhibition or induction respectively. Notch signaling is also used in an iterative way to regulate consecutive cell fate decisions required for the formation of specialized tissues. Signaling is initiated when transmembrane Notch ligands, belonging to the Delta–Serrate–Lag2 family, bind to Notch receptors. Upon stimulation, the receptor is proteolitically processed by the sequential actions of two enzymes: tumor necrosis factor converting enzyme and γ-secretase, leading to the release of the Notch intracellular domain (NICD), which translocates to the nucleus and interacts with CBF1/SuH/Lag1 (CSL) transcription factors to initiate expression of target genes, including the basic helix-loop-helix Hes and Hey genes (Bray 2006).
Notch signaling and the generation of β-cells
Notch signaling is important for regulating the expression of neurogenin 3 (Ngn3 or Neurog3), a critical transcription factor for the formation of pancreatic endocrine cells. Ngn3-expressing cells are considered to be islet progenitors (Apelqvist et al. 1999, Gu et al. 2002), and lack of Ngn3 expression leads to complete ablation of the endocrine component in developing mice (Gradwohl et al. 2000). Notch signaling represses transcription of Ngn3 through Hes activation to prevent premature endocrine differentiation. This step serves to expand the pool of pancreatic progenitors before differentiation is initiated and notch inhibition results in Ngn3 expression, and further differentiation towards the endocrine fate (Apelqvist et al. 1999, Jensen et al. 2000). Consistent with this, mouse embryos overexpressing Ngn3 or lacking the CSL or Delta-like1 mediators or both Hes1 alleles show an accelerated endocrine differentiation coupled with pancreatic hypoplasia (Apelqvist et al. 1999, Jensen et al. 2000). Conversely, sustained notch activity strongly represses endocrine differentiation (Hald et al. 2003, Murtaugh et al. 2003).
Modulators of the Notch signaling pathway
Given its role in pancreas development, repressing the notch pathway in vitro would be expected to enhance differentiation towards a β-cell fate. Notch repressors could be employed after Hedgehog downregulation and subsequent Pdx1 activation, in accordance with the known developmental process in vivo. However, in three studies employing hESCs that incorporated notch inhibition in their differentiation protocols (D'Amour et al. 2006, Phillips et al. 2007, Cho et al. 2008), this inhibition had only a slight impact on promoting endocrine differentiation. Both of these studies used N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT), which was added after Pdx1 activation. DAPT belongs to a class of compounds called γ-secretase inhibitors, which, as the name suggests, inhibit notch signaling by preventing activation of the γ-secretase enzyme which, in turn, blocks NICD release (De Strooper et al. 1999). However, not all γ-secretase inhibitors specifically affect notch signaling (Lundkvist & Näslund 2007), and γ-secretase inhibitors other than DAPT may prove to be more efficient within the context of β-cell differentiation. The first γ-secretase inhibitors, including the peptidyl aldehydes MG-132 and MDL-28170, were identified more than a decade ago (Higaki et al. 1995, Kopan et al. 1996). Soon thereafter, a difluoroketone chemical, named MW167 (sometimes referred to as compound 1), was also shown to inhibit γ-secretase activity (Wolfe et al. 1998). All of these compounds also tested positive for Notch inhibition, either in vivo (Taniguchi et al. 2002) or in vitro (Beck & Slack 2002). More recent inhibitors that are also known to modulate Notch include a hydroxyethylene peptidomimetic named L-685,458 (Shearman et al. 2000, Sernee et al. 2003) together with its homologous counterpart III-31-C (Li et al. 2000, Esler et al. 2002), the structurally related compounds D and E (Seiffert et al. 2000, Beher et al. 2001), CBAP (Beher et al. 2001), LY411575 (Dovey et al. 2001, Wong et al. 2004), a dibenzazepine and benzodiazepine (Milano et al. 2004), and a group of epoxides consisting of compounds IL, ILX, and KILX (Piper et al. 2003). The abundance of pharmacological inhibitors of Notch signaling makes this another attractive target for inclusion in future in vitro differentiation protocols, as shown in Fig. 1.
Other modulators
A number of biologically active compounds that have been used in published endocrine pancreas differentiation protocols cannot be classified under the ‘signaling pathway’ scheme described in this review, either because they operate through alternative pathways or because their mode of action is uncertain.
Glucagon-like peptide-1 (GLP-1) agonists are thought to promote β-cell differentiation by acting through several intracellular pathways such as the PI3K, the Hedgehog, the Ras/MAPK, the PKA, and the MEK/ERK pathway to modify the expression of β-cell transcription factors, including Pdx1 (Buteau et al. 1999, Montrose-Rafizadeh et al. 1999, Wang et al. 1999, Zhou et al. 2002, Hui et al. 2009). Recent studies have shown that the GLP-1 peptide and exendin (a potent GLP-1 agonist) can increase the percentage of insulin-expressing cells in mouse (Ku et al. 2004, Bai et al. 2005), rhesus monkey (Lester et al. 2004), and hESC pancreatic differentiation protocols (Yue et al. 2006, Cho et al. 2008, Hui et al. 2009, Zhang et al. 2009). Insulin-like growth factor 2 also has a known role in β-cell neogenesis and islet hyperplasia (Petrik et al. 1999, Calderari et al. 2007), and has been used at late stages of the differentiation process in the studies of β-cell generation from hESCs (Jiang et al. 2007a, Phillips et al. 2007).
Retinoic acid is essential for pancreatic development through the induction of Pdx1 expression (Martín et al. 2005, Molotkov et al. 2005) and promoting the generation of Ngn3+ endocrine progenitors (Öström et al. 2008). Consistent with this, mouse (Micallef et al. 2005, Nakanishi et al. 2007) and human (D'Amour et al. 2006, Jiang et al. 2007b, Shim et al. 2007, Kroon et al. 2008, Johannesson et al. 2009, Zhang et al. 2009) ES cell pancreatic differentiation protocols that incorporate retinoic acid result in the induction of a pancreatic endoderm-like stage.
Some biologically active molecules have been used in ES differentiation protocols with little insight on their mechanisms of action, including indolactam V, nicotinamide, sodium butyrate, and β-cellulin. Indolactam V is thought to promote differentiation at least, in part, by activating protein kinase C signaling. It was reported to induce expression of Pdx1 from mouse and hESCs, and this effect was enhanced in the presence of FGF10 (Chen et al. 2009). Nicotinamide has been reported to promote endocrine differentiation in fetal pancreatic cells (Otonkoski et al. 1993), and has been widely used in in vitro differentiation protocols, either to induce pancreatic differentiation in undifferentiated mESCs (Chen et al. 2008, Vaca et al. 2008) or, more commonly, as a ‘maturation factor’ in the later stages of mouse (Hori et al. 2002, Vaca et al. 2003, Ku et al. 2004, Naujok et al. 2008a,b) and hESC pancreatic differentiation protocols (Jiang et al. 2007a,b, Cho et al. 2008, Zhang et al. 2009). The precise mode of action of nicotinamide as a morphogen is uncertain, but some of its effects may be mediated through the inhibition of poly(ADP-ribose) synthase which is thought to cause chromatin rearrangements and changes in gene transcription (Otonkoski et al. 1993). β-Cellulin has been used in a study by Cho et al. (2008), where it was reported to act synergistically with nicotinamide to sustain Pdx1 expression and induce pancreatic endoderm and islet maturation. Its effects might be due to activation of the epidermal growth factor pathway (Ishiyama et al. 1998).
Finally, a number of biologically active molecules have been shown to influence pancreatic differentiation but have not yet been incorporated into protocols to drive ES cells to an endocrine pancreas phenotype. One example is stauprimide, which has been shown to promote a DE fate in both mouse and hESCs by interacting with the NME2 protein, causing c-Myc downregulation (Zhu et al. 2009). Another example is conophylline, a vinca alkaloid (Umezawa et al. 1994) that has been reported to promote differentiation of murine bone marrow mesenchymal cells (Hisinaga et al. 2008) as well as rat pancreatic acinar carcinoma and pancreatic precursor cells (Umezawa et al. 2003, Ogata et al. 2004, Kitamura et al. 2007) into insulin-producing cells. Its effects are possibly due to induction of the Ngn3 transcription factor by a p38-dependent mechanism (Umezawa et al. 2003). Another compound, trichostatin A (TSA), which is a hydroxamic acid histone deacetylase inhibitor (HDACi), has been shown to promote differentiation in human endometrial adenocarcinoma cells (Uchida et al. 2005) and promyelocytic leukemia cells (Kitamura et al. 2000), and to induce insulin expression in mouse bone marrow stem cells (Tayaramma et al. 2006). Similarly, other hydroxamic acid HDACis, such as SAHA and pyroxamide, have been shown to induce terminal differentiation in various cell lines including murine erythroleukemia cells (Butler et al. 2001), human endometrial adenocarcinoma cells (Uchida et al. 2005), and human breast cancer cells (Munster et al. 2001). Sodium butyrate has HDACi activity (Rada-Iglesias et al. 2007) which may account for its reported effects to induce DE formation from mouse and hESCs respectively (Goicoa et al. 2006, Jiang et al. 2007b). HDACis have also been reported to upregulate gelsolin, a protein that is a major building block for actin filament formation and which may be associated with the pro-differentiating effects of this class of compound (Mielnicki et al. 1999, Huang & Pardee 2000, Rombouts et al. 2002, Glaser et al. 2003). Histone deacetylases have been implicated recently in the regulation of expression of key pancreatic transcription factors (Haumaitre et al. 2008), and HDACis, such as TSA and sodium butyrate, have been shown to influence the timing and determination of pancreatic cell fate (Haumaitre et al. 2008), suggesting that this class of compounds may be useful in in vitro differentiation protocols.
Summary and conclusions
A number of recent studies have demonstrated that recapitulating in vitro the signals controlling the development of the endocrine pancreas in vivo offers a promising strategy for generating insulin-expressing β-cells from pluripotent ES cells. However, current protocols are not yet optimized for a number of reasons, including the pleiotropic effects induced by individual morphogens, the complexity of the signaling pathways involved, and the likelihood of crosstalk between pathways. We suggest that the establishment of efficient and reproducible protocols will be facilitated by focusing on the intracellular signaling pathways that regulate and direct the developmental transitions. Figure 1 shows a proposed differentiation protocol based on known developmental processes and on currently available pharmacological modulators of the signaling systems. Initially, ES cells can be driven to differentiate into mesendoderm by suppressing the PI3K pathway and keeping Nodal and Wnt signaling sufficiently high. DE can then be generated by maintaining suppression of the PI3K pathway while activating the Nodal pathway activation. Thereafter, developmental evidence is in favor of suppressing the Wnt pathway for generating posterior foregut endoderm, followed by repression of Hedgehog signaling to allow for specification of the pancreatic domain, while inhibiting Notch signaling to promote further differentiation towards the endocrine fate. Finally, suppressing PI3K and activating Hedgehog signaling might further enhance the differentiation towards the much desired functionally mature β-cells.
Declaration of interest
Neither author has any conflict of interest concerning the impartiality of this review article.
Funding
The generation of this review article did not receive any grant support from any funding agency in the public, commercial, or not-for-profit sector.
- Received in final form 7 April 2010
- Accepted 12 April 2010
- © 2010 Society for Endocrinology
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










