- Made available online as an Accepted Preprint 9 January 2009
- Accepted Preprint first posted online on 9 January 2009
Genome-wide identification of DNA–protein interactions using chromatin immunoprecipitation coupled with flow cell sequencing
- Department of Cancer Endocrinology, BC Cancer Research Center, 675 West 10th Avenue, Vancouver, BC, Canada V5Z 1L31Micheal Smith Genome Sciences Centre, BC Cancer Agency, Suite 100-570 West 7th Avenue, Vancouver, BC, Canada V5Z 4S6
- (Correspondence should be addressed to S J M Jones; Email: sjones{at}bcgsc.ca)
Abstract
The transcriptional networks underlying mammalian cell development and function are largely unknown. The recently described use of flow cell sequencing devices in combination with chromatin immunoprecipitation (ChIP-seq) stands to revolutionize the identification of DNA–protein interactions. As such, ChIP-seq is rapidly becoming the method of choice for the genome-wide localization of histone modifications and transcription factor binding sites. As further studies are performed, the information generated by ChIP-seq is expected to allow the development of a framework for networks describing the transcriptional regulation of cellular development and function. However, to date, this technology has been applied only to a small number of cell types, and even fewer tissues, suggesting a huge potential for novel discovery in this field.
Introduction
The transcriptional networks driving mammalian cell development and function are only beginning to be elucidated. In many tissues transcription factors critical to normal development and function have been identified but, in general, only a handful of their direct targets are known. Next-generation, or flow cell sequencing technologies are quickly becoming the standard for numerous genomic analyses (Mardis 2007, 2008, Down et al. 2008, Holt & Jones 2008, de Hoon & Hayashizaki 2008, Marioni et al. 2008, Morozova & Marra 2008, Mortazavi et al. 2008, Schuster 2008, Wold & Myers 2008). In particular, the extreme quantity of short-read sequence information that can be generated by these technologies matches extremely well with the analysis of DNA fragments enriched by chromatin immunoprecipitation (ChIP-seq; Johnson et al. 2007, Mardis 2007).
ChIP is now standard practice in the identification of histone modification locations and transcription factor binding sites (Elnitski et al. 2006, Wu et al. 2006, Collas & Dahl 2008, Massie & Mills 2008). For this purpose, cells are typically initially treated with a cross-linking agent, often formaldehyde, to covalently link DNA-binding proteins to chromatin (Fig. 1). Next the cells are lysed, and genomic DNA is isolated and subsequently sonicated to produce sheared chromatin. An antibody specific to the protein of interest is then added to the sonicated material and DNA fragments bound to the protein of interest isolated via immunoprecipitation. DNA fragments are then released by reversing the cross-links and the fragments purified.
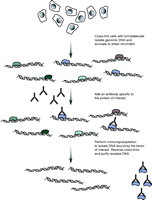
An overview of the chromatin immunoprecipitation (ChIP) procedure. Cells are initially treated with a cross-linking agent that covalently links DNA-interacting proteins to the DNA. The genomic DNA is then isolated and sheared, typically by sonication, into a suitable fragment size distribution (100–300 bp is typically used for ChIP-seq). An antibody that specifically recognizes the protein of interest is then added and immunoprecipitation used to isolate appropriate protein–DNA complexes. The cross-links are then reversed and the DNA fragments purified.
Classically, the DNA obtained from ChIP reactions is assessed by PCR. For this method primers flanking regions of interest are used, and PCR products from ChIPs using the protein-specific antibody are compared with products from IgG control samples, with higher levels of PCR product in protein-specific antibody ChIPs indicating enrichment of the target region. An advancement on this technique is the use of quantitative real-time PCR to more accurately assess the enrichment levels of interrogated sites. A limiting factor for such PCR-based assays is that they are site specific and only provide information on regions identified a priori.
To allow for the discovery of novel sites, researchers began hybridizing material obtained by ChIP to genomic microarrays, and particularly to cost-effective promoter arrays (ChIP–chip; Wu et al. 2006, Collas & Dahl 2008, Massie & Mills 2008). More recently, various strategies have been developed to sequence concatenated fragment ends from the chromatin-immunoprecipitated material using Sanger sequencing, such as ChIP-serial analysis of chromatin occupancy (Impey et al. 2004), ChIP-serial analysis of binding elements (Chen & Sadowski 2005), ChIP-sequence tag analysis of genomic enrichment (Bhinge et al. 2007), and genome-wide mapping technique (Roh & Zhao 2008), as summarized in Table 1. Although these methods overcome the bias inherent in the use of microarrays, the generation of sufficient sequence reads to identify all but the most significant of sites is highly cost-prohibitive. Subsequently, the use of parallel pyrosequencing was explored, via the Roche 454 DNA sequencer to identify binding sites for p53 (Wei et al. 2006), Oct4 and Nanog (Loh et al. 2006), and estrogen receptor alpha (Lin et al. 2007) by ChIP-PET. This method though lacked the throughput necessary to cost-effectively survey a mammalian genome at a satisfactory level of redundancy, limiting its ability to discriminate a truly comprehensive set of enriched regions. To further improve upon this method, researchers began exploiting short-read flow cell sequencing to identify immunoprecipitated DNA fragments (ChIP-seq; Barski et al. 2007a, Johnson et al. 2007, Robertson et al. 2007). These, and subsequent reports, have made it clear that the future of the global identification of histone modifications and binding sites of DNA-interacting proteins, such as transcription factors, currently lies in the use of this technique.
Summary of techniques for the genome-wide analysis of chromatin immunoprecipitation-enriched DNA
Advantages of ChIP-seq
ChIP–chip and ChIP-seq are currently the two main competing technologies for the genome-wide identification of chromatin-immunoprecipitated material. However, ChIP-seq has a number of key advantages over ChIP–chip (Wu et al. 2006, Collas & Dahl 2008, Massie & Mills 2008), premier among these are that ChIP-seq is relatively unbiased and truly genome wide. In fact, most available microarray designs represent a limited number of sites that only represent a fraction of the total genome. Although Affymetrix and NimbleGen do produce whole-genome tiling array sets for several species, the use of these arrays has been limited for several reasons. First, the cost of these array sets is significant, and although the initial investment in sequencing instrumentation and the cost of reagents for ChIP-seq mean that the expense of a ChIP-seq experiment can also be considerable, they are more than competitive with ChIP–chip. Also, the availability of sequencing centers or commercial facilities for outsourcing sequencing removes the upfront costs of purchasing the required instrumentation. In spite of this, ChIP-seq is a nascent technology and it is anticipated that costs will decrease while sequence throughput and access to the technology will increase in future. Another issue with ChIP–chip is that it is currently necessary to use multiple arrays to survey a whole genome; in fact, one recent report used a total of 37 arrays to survey the mouse genome (Barrera et al. 2008). Also, the dynamic range and signal-to-noise ratio in ChIP–chip experiments are limiting, and cross-hybridization between probes can also play an obfuscating role (Johnson et al. 2007, Mardis 2007, Massie & Mills 2008). Another consideration is the size and spacing of the probes spotted on the array. Many custom arrays use probes several hundred base pairs in length or longer. This can make the identification of the actual binding site within the probe difficult, especially for transcription factors with poorly characterized binding site preferences or that bind highly degenerate sequences. Even state-of-the art commercial arrays (Qi et al. 2006) cannot offer the spatial resolution possible with ChIP-seq. In fact, using ChIP-seq, the actual binding site of a factor can often be identified within 10–30 bp of the peak maximum (Kharchenko et al. 2008, Zhang et al. 2008a), with even greater precision possible with greater levels of sequence information, better analysis methods, and longer sequence read lengths. Another important consideration is the amount of input material required. ChIP–chip experiments require upwards of 4–5 μg of material; although whole-genome amplification can be used to amplify ChIP-enriched DNA, this can lead to increased background, the possibility of poor, or no amplification of some target regions, as well as various other artifacts (O'Geen et al. 2006). On the other hand, ChIP-seq requires as little as 10 ng, making it the clear choice when sample input is limiting.
Flow cell sequencing platforms for ChIP-seq
The first flow cell sequencing device on the market was the genome sequencing device from 454 Life Sciences (now owned by Roche). Subsequently, Solexa (now Illumina) released their genome analyzer. Currently, Applied Biosystems and Helicos also offer alternatives. The Illumina device is currently the most commonly used, due to its availability, time in the market, and as it has several advantages over the 454 for the analysis of chromatin-immunoprecipitated DNA. Specifically, the Illumina device generates over ten times the number of DNA sequences, albeit shorter, at roughly a tenth the cost of the 454 (von Bubnoff 2008). All next-generation sequencing devices have demonstrated rapid evolution in their technology and performance. Currently, the Illumina device can produce reads of 75–100 bases, while the 454 can produce sequence reads in excess of 500 bases. For ChIP applications, the shorter reads generated by the Illumina device are typically of sufficient length for the majority of sequence reads to be uniquely mapped to the genome, and the increased data quantity, for the same cost, is significant.
Flow cell sequencing methods, unlike Sanger sequencing, sequence DNA fragments in parallel (Fig. 2). Using this method, current Illumina devices can sequence tens of millions of DNA fragments in parallel and produce over 10 Gbp of data in a single run. In Illumina sequencing, adapters are first ligated to size-selected DNA fragments and used to prime low-cycle PCR amplification of the DNA. PCR products are then attached to a flow cell and ‘bridge’ amplification used to generate clonal clusters of identical DNA products. A sequencing primer homologous to the ligated adapters is then annealed, and used to initiate a sequence by synthesis process using reversibly terminated fluorescently labeled nucleotides. After each cycle of nucleotide addition, fluorescence microscopy is used to capture an image of the flow cell. When the run is complete, image analysis is performed to generate base calls (Bentley et al. 2008).
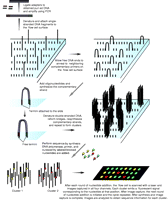
An overview of the sequence by synthesis method. Initially, adapters are ligated to the purified DNA fragments and low-cycle PCR used to amplify the DNA. The amplified DNA is subsequently denatured to form single-stranded products and one end attached to the surface of a flow cell. Subsequently, the free end is allowed to anneal to a primer complementary to one of the adapters that is attached to the flow cell surface. This primer is then used to prime synthesis of the complementary strand. The now double-stranded bridge structures are then denatured producing two single-stranded products and this cycle is repeated to form clusters. After cluster formation, the sequencing phase of the process begins with the addition of DNA polymerase, primer, and each of the nucleotides that are fluorescently labeled with different fluorophores and are blocked to prevent extension. This results in the addition of one nucleotide to each DNA fragment. Fluorescence microscopy is then used to capture images of the flow cell. After imaging, the oligonucleotides on the cell are deprotected and the next round of synthesis is initiated. After the process is complete, image analysis is used to determine the sequence of the DNA fragments in each cluster.
Regardless of the device used for ChIP-seq, the sequences obtained are initially computationally mapped back to the reference genome by a sequence alignment algorithm (Li et al. 2008a). ChIP-seq relies on DNA regions enriched by the ChIP being sequenced more often than would occur by chance. Thus, sequence alignments are used to identify over-represented regions that form ‘peaks’ that mark the location of DNA–protein interactions.
Applications of ChIP-seq
Mapping of chromatin modifications
One of the major applications of ChIP-seq is in the genome-wide mapping of histone modifications. In fact, the first reported use of ChIP-seq was to identify the genome-wide locations of 20 different histone lysine and arginine methylations in addition to H2A.A, RNA pol II and CTCF in human CD4+T cells (Barski et al. 2007a). Previous ChIP–chip studies reported that H3K4 methylation was a hallmark of active genes (Heintzman et al. 2007) and that, inversely, H3K27 methylation was a hallmark of repressed genes (Lee et al. 2004, Bernstein et al. 2005, Kim et al. 2005, Roh et al. 2005, 2006, Boyer et al. 2006). Of interest, genes with so-called bivalent marks were also identified, in which the opposing H3K4me3 and H3K27me3 marks are present at the same site (Bernstein et al. 2006, Roh et al. 2006). However, these ChIP–chip studies suffered from limited genome coverage and resolution (Bernstein et al. 2007). The advent of ChIP-seq was not only able to confirm these results, but also allowed the association of unique modifications with gene activation or repression in a cost-effective, high resolution, and truly genome-wide scale (Barski et al. 2007a). Also using CD4+T-cells, Wang et al. (2008) further mapped 18 different histone acetylations using ChIP-seq. Combining their data with Barski's, they identify a ‘modification module’ of 17 co-occurring histone modifications associated with genes with high expression, and suggest that these histone modifications act cooperatively to prepare chromatin for transcriptional activation.
ChIP-seq has also been used to map the locations of the H3K4me3, H3K27me3, H3K9me3, H3K36me3, and H4K20me3 histone modifications in mouse ES cells, neural progenitor cells, and embryonic fibroblasts (Mikkelsen et al. 2007). This study pointed toward the resolution of bivalent H3K4me3/H3K27me3-marked genes in progenitor cells, into expressed genes marked only with H3K4me3, and repressed genes marked only with H3K27me3, in committed cell types. More recently, ChIP-seq further allowed the validation of H3K4me1 as a mark of distal enhancer regions in HeLa cells and mouse liver (Robertson et al. 2008).
Furthermore, several interesting examples of the utility of ChIP-seq-derived histone modification data have been reported. For example, using the data of Barski et al. (2007a) and Hon et al. (2008) developed an unsupervised method called ChromaSig for the identification and clustering of histone mark profiles. From this method, they identified 16 different commonly occurring histone modification patterns. For example, H3K4me1, H3K4me2, H3K4me3, H3K9me1, H2AZ, H4K20me1, and RNA polymerase II were found to commonly co-occur. These marks are all classically associated with active genes and this cluster was commonly localized to 5′ ends of RefSeq transcripts and in conserved regions. Also, using the data of Barski et al. (2007b) and Schmid & Bucher (2007), were able to map nucleosome positions with a high degree of precision. They identified a clear peak periodicity for several of the histone variants and used this to precisely identify nucleosomes centered at +120, 300, and 480 bp from transcriptional start sites (TSS). Upstream from TSS, the closest nucleosome was identified at −180 bp from the transcriptional start site. In response, Barski and colleagues noted that although ChIP-seq data can be used to map nucleosome positions, it is limited in only identifying nucleosomes that have the modifications assessed, although antibodies against core histone could be used to overcome this (Pokholok et al. 2005).
Mapping transcription factor binding sites
The other primary use of ChIP-seq is the global identification of transcription factor binding sites. In one of the initial studies utilizing the massively parallel sequencing capacity of the Illumina sequencer, Johnson et al. (2007) identified 1946 binding sites for the transcription factor, neuron-restrictive silencing factor (NRSF) in Jurkat cells. The data were determined to be of high specificity and sensitivity; and to be highly comprehensive, as most high-affinity NRSF sites in the genome were occupied. Motif finding, on the obtained peaks, confirmed the known NRSF binding motif, and further demonstrated that the two half sites in the canonical motif can have variable spacing. Interestingly, many genes critical to pancreatic function were bound by NRSF, although the functional relevance of this remains to be determined, given that these sites were identified in a T-cell cell line. Shortly thereafter, Robertson et al. (2007) described the identification of binding sites for STAT1 in HeLa S3 cells in an unstimulated and IFN-γ-stimulated state. Over 40 000 STAT1 sites were identified in the stimulated cells, while 11 004 were identified in unstimulated cells. This study highlighted the dramatic changes that can occur in a transcription factor binding site repertoire in response to external stimuli. Interestingly, subsequent reanalysis of these data in the context of ChIP-seq-determined H3K4me1 and H3K4me3 profiles, which are associated with active genes, revealed that the majority of the STAT1 binding sites, which were unique to stimulated cells, were already associated with H3K4me1 and H3K4me3 in unstimulated cells (Robertson et al. 2008). Thus indicating that activated STAT1 binding is heavily influenced by the pre-existing chromatin context.
A study by Chen et al. (2008) used ChIP-seq to map the binding sites of 13 transcription factors as well as two transcriptional regulators in mouse ES cells, identifying between 1126 and 39 609 sites for each factor. This study provided a great deal of insight into the co-occurrence of specific sets of transcription factors in ES cells, for example NANOG, SOX2, OCT4, SMAD1, and STAT3 in so-called ‘ES enhanceosomes’. More recently, a study by Marson et al. (2008) which also profiled the binding sites of OCT4, SOX2, NANOG, as well as TCF3 in mouse ES cells, identified 14 230 site co-occupied by all four factors. These data, in combination with H3K4me3 location data, were then used to identify regions active in the regulation of miRNA genes.
Nielsen et al. (2008) used ChIP-seq to assess PPARγ, retinoid X receptor (RXR), and Pol II binding sites in a model of adipocyte differentiation. For this purpose, 3T3-L1 cells were induced to differentiate and cells were harvested for ChIP at days 0, 1, 2, 3, 4, and 6. In this study, the number of PPARγ sites increased dramatically with differentiation, while the number of RXR sites also increased, although less dramatically. On day 6, 5236 overlapping PPARγ:RXR sites were identified, the majority of which were found within introns. They also found that overlapping sites were enriched at genes with increased Pol II occupancy, with particularly high levels of occupancy near genes associated with glucose and lipid metabolism. Using de novo motif analysis on the identified peak sequences, they identified a motif similar to the C/EBP position weight matrix.
ChIP-seq has also proven of value in the identification of transcription factor binding sites using tissue samples. Wederell et al. (2008) mapped the binding sites of FOXA2 in the adult liver, identifying over 11 000 FOXA2-binding sites. From this, they found that 43.5% of liver-expressed genes have an associated FOXA2-binding site. Numerous enriched motifs were identified associated with FOXA2-binding sites, including those for HNF4α and HNF1α, which are known to cooperate with FOXA2 in liver development (Odom et al. 2006).
ChIP-seq considerations
Antibody selection
One of the limiting factors in ChIP-based studies is the availability and identification of suitable antibodies. In ChIP-seq, all of the material that is immunoprecipitated is sequenced and used in peak building. As such, cross-reactive antibodies will generate peaks from the binding of non-targeted proteins, while antibodies with insufficient affinity will be incapable of precipitating sufficient material to produce a good signal-to-noise ratio (Wang et al. 2008). In principle, ChIP-seq should be able to overcome some of the shortcomings of antibodies with low affinity but high specificity, as deeper sequencing can allow regions with weak enrichment to become statistically significant. In any event, the standards for antibody quality, and in particular antibody specificity, are higher for ChIP-seq than for the interrogation of specific sites by ChIP-PCR or ChIP-qPCR. Given this, researchers should be skeptical of the utility of an antibody that is claimed to be of ChIP quality for use in ChIP-seq particularly when this claim refers to the purity grade of the antibody. In spite of this, for use in ChIP-seq experiments, it is advisable that, as a general ‘rule of thumb’, less than 20% of the total protein bound by an antibody be from cross-reactive species in western blot analysis of cell lysates from the cell type of interest (based on ENCODE project standards; Synder, personal communication, November 30, 2008). Also, it is advisable that ChIP-qPCR analyses be performed on several known targets and consistently given a minimum of 20-fold enrichment, when compared with levels in IgG ChIP reactions, and that several negative sites are, likewise, not enriched. It is also advisable to use only antibodies that immunoprecipitate enough ChIP material to produce a visible smear after EtBr or SYBR green staining in the 100–300 bp range when run on a PAGE gel.
One issue to keep in mind is that some antibodies will only recognize factors that are in specific conformations, or only in the absence of cofactors that obscure the antibody recognition site. For these reasons, where possible, it is advisable to use a mixture of antibodies or to perform replicate ChIP-seq experiments using different antibodies to the same factor. For example, to identify CTCF-bound sites in the human genome by ChIP–chip, a mixture of nine separate monoclonal antibodies was used (Chen et al. 2007). However, in most cases, this is not practical, largely due to the current scarcity of ChIP-seq quality antibodies.
It is also worth noting that in the absence of suitable antibodies it is possible to generate transgenic or knock-in cell lines or mouse strains that express the factor of interest fused with a suitable tag or tandem affinity tag (Zhou et al. 2004). With these techniques though artifact peaks are a concern and appropriate controls are essential. In spite of this, it is clear that there are numerous currently available antibodies to transcription factors and their cofactors that are useful for ChIP-seq; it just remains for researchers to identify them. Furthermore, the development of new high-quality antibodies to mammalian transcription factors appears to be an area of active investigation by several companies, and, in the future, it is expected that a greater number of appropriate antibodies will become available.
The identification of enriched sites
To identify enriched regions that represent binding sites from a ChIP-seq experiment, the obtained short sequence reads from a sequencing run are first aligned to the genome (Fig. 3), as such, it should be noted that ChIP-seq is a resequencing technology and is therefore restricted to use with organisms whose genomes have been sequenced. In any event, a series of non-identical sequence reads will cluster together in locations where DNA was bound by the protein of interest, allowing the identification of sites enriched above background. The number of reads present in these clusters depends on the level of enrichment at the site, which typically correlates with the level of occupancy of the site in the case of a transcription factor, or the extent of the modified region for histone marks. It should be stressed that ChIP is an enrichment and not a purification strategy, and, typically, only a few percent of the sequenced reads fall within identified peaks. It should also be kept in mind that some regions may appear enriched that do not interact with the protein of interest, for example regions of the genome predisposed to fragmentation. This is likely influenced by factors such as repetitive elements and the level of openness of the chromatin. Incompleteness in the reference genome and natural polymorphisms, especially copy number variation, will also play a role in generating false peaks.
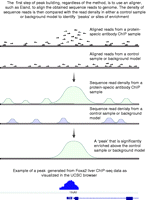
A schematic depicting how peaks are formed from sequence reads. For peak identification, sequence reads are first aligned to the genome. Regions of protein–DNA interaction will have an enriched concentration of reads when compared with the background model. The background read density is either obtained from a control sample or computationally predicted in the absence of a control. Thus, tag density can be used to identify peaks, or sites of enrichment, that correspond to locations of interaction between the protein of interest and the genomic DNA. An example of a peak, visualized using the UCSC browser, indicating a FOXA2-binding site in the promoter of the transthyretin (Ttr) gene found in the liver.
Although several ChIP-seq experiments have used random background models to identify regions enriched above random expectation, the use of empirically derived control samples, preferentially from the same cell type, is an increasingly important consideration. Various methods of deriving control samples have been proposed, including the use of sonicated input DNA or material from species-specific IgG immunoprecipitations. The use of such controls typically allows the discovery of a greater number of enriched regions, with lower levels of false positives. For experiments without a matching control, many false positives can be removed by filtering out peaks that overlap repetitive regions. Even still, one study noted 96 vs 83% concordance between peaks obtained for OCT4 and NANOG when the data were processed with and without a control (Ji et al. 2008). Thus, although it is clear that experiments that do not use controls can still be successful, this is at the expense of some level of specificity, and this needs to be weighed against the cost of generating the control sample.
The development of statistical approaches for the identification of enriched regions has proven to be an active area of investigation with several tools for identifying peaks being published, including FindPeaks 3.1, F-Seq, SISSR, QuEST, MACS, the ChipSeq Peak Finder, ChIPDiff, CisGenome, and the ChIP-seq processing pipeline (Johnson et al. 2007, Boyle et al. 2008, Fejes et al. 2008, Jothi et al. 2008, Kharchenko et al. 2008, Valouev et al. 2008, Xu et al. 2008, Zhang et al. 2008a,b). In general, these algorithms function in a similar manner. In the absence of a control, a background model is used to identify enriched regions, with Poisson (Zhang et al. 2008a), Monte Carlo (Fejes et al. 2008), negative binomial (Ji et al. 2008), and other (Kharchenko et al. 2008) background models being proposed. When a control is available, peaks are typically named based on the number of reads in a region, or window, when compared with the number of reads in the same region in the control (Ji et al. 2008, Jothi et al. 2008, Valouev et al. 2008, Zhang et al. 2008a). Regardless of the method used, FDR estimates are based on the level of enrichment at the site, either globally or locally, compared with the background model used. The relative advantages and disadvantages of these different algorithms remain to be determined and, despite these advances, ChIP-seq is a relatively immature technology and appropriate informatic tools for the identification of enriched sites are still being developed.
With current tools, the choice of a significance or enrichment threshold to discriminate real binding sites from background is often not clear and users must make a choice based on calculated false discovery rates and/or on the level of enrichment of the expected binding motif. Threshold choice is especially difficult for transcription factors, as most bind at low affinity to a large number of sites, and ‘saturation’, i.e. a plateau in the number of enriched regions with increasing sequencing depth, is typically not obtained, unlike with histone modification studies where saturation typically is achieved (Robertson et al. 2008). This makes the discernment between a ‘true’ low-affinity site and background particularly difficult. As such, transcription factor ChIP-seq experiments cannot, in general, claim to be comprehensive. Despite this, the functional relevance of such low-affinity sites is uncertain, as discussed in greater detail later in this review, and thus the relevance, or even desirability, of obtaining saturation is questionable.
Associating peaks with genes
To date, standard practice for associating peaks with genes has been to use some distance criteria from TSS or from the gene unit. For example, Johnson et al. (2007) mapped peaks to genes if the peak was within ±20 kb of the genes TSS, while Wederell et al. (2008) mapped peaks to genes if the peak was within −10 kb from the TSS to +1 kb from the transcriptional termination site. Chen et al. (2008) used a more sophisticated method, and determined the distribution of distances from known TSSs to the peaks produced by each factor assessed. They then determined where these distributions deviated above background expectation. Peaks were then associated with the nearest gene, within this threshold, and each peak–gene association scored based on its significance above random expectation. Despite these efforts, complications clearly arise. In part, this is due to gene-dense regions where a peak may be in close proximity to several different genes. In these situations, the closest gene is not necessarily the gene under regulation, and a transcription factor may in fact influence several of the genes surrounding it. Clearly, better association metrics that take into account the likelihood of a site–gene association being correct need to be developed, and this continues to be an active area of research. Chromosome conformation capture (3C), and its higher throughput extensions, chromosome conformation capture-on-chip (4C), and chromosome conformation capture carbon copy (5C), and other extensions of these approaches that detect interactions between genomic loci and identify cis-regulatory sites associating with TSS during transcriptional initiation stand to help resolve these issues (Dekker et al. 2002, Dostie et al. 2006, Simonis et al. 2006, Zhao et al. 2006).
Determining the functional relevance of identified sites
Perhaps one of the first questions one might have of ChIP-seq data, particularity for a transcription factor, is ‘how do I determine if the thousands of sites I have identified are functionally relevant?’. To answer this question is a complex issue, specifically in the context of knowing where to set thresholds, as mentioned above.
It has been suggested that many transcription factor binding sites identified by ChIP-chip in Drosophila blastoderm are non-functional (Li et al. 2008b), as many of the identified sites were low scoring against known binding motifs, adjacent to inactive genes, distant from transcribed genes, or in protein-coding regions. Additionally, in mammalian systems, nearly half of identified binding sites are associated with inactive genes (Hatzis et al. 2008, Lupien et al. 2008, Wederell et al. 2008). However, in our experience, no distinction in binding affinity can be made between sites associated with active versus inactive genes, or distal versus proximal sites. The possible lack of functionality of these sites becomes even less clear when the biology of the binding factor is taken into consideration. For example, many transcription factors either require cofactors for activity or can actively repress genes. Thus, these sites may represent functional repression, or may be functional in a different cellular context in which appropriate cofactors are expressed.
Despite this, several methods can be used to provide an argument that sites are functional. For example, the expression of genes with or without an associated site can be compared (Johnson et al. 2007, Chen et al. 2008). This can be convincing if the biology of the factor is straightforward, i.e. the factor is a strong activator or a strong repressor. However, this is often not the case, and, often, factors can act as both activators and repressors within the same cell depending on the context of the site, or have varying levels of effect, depending on recruited cofactors. Another method is to assess the expression of target genes, in cells in which the factor of interest has been knocked out or suppressed. In this case, functionally relevant sites should be revealed by the alteration in expression of the associated target gene. A concern with these studies is the compensation of the factor by a related family member. A third method is to look for concordance of binding sites and histone modifications that demarcate enhancer and promoter regions, such as H3K4me1 and H3K4me3 (Barski et al. 2007a, Mikkelsen et al. 2007, Robertson et al. 2008). Marson et al. (2008) used this approach to identify active promoter and enhancer regions for miRNA genes. Comparing identified FOXA2 sites in the liver and STAT1 sites in stimulated Hela cells, Robertson et al. (2008) demonstrated that 84% and 87% of these sites were associated with H3K4me1 or H3K4me3 marks respectively. Although the extent to which H3K4me1 and H3K4me3 do predicate function is unknown, these data suggest that most identified transcription factor binding sites are associated with regulatory regions and are likely functional, although perhaps only in the right physiological context (Wederell et al. 2008).
A more direct method to prove that a site is functional is to demonstrate that it alters the activity of a reporter gene. This demonstrates that a genomic fragment containing the identified binding site or sites is active; however, without specifically mutating or altering the binding site for the factor of interest, it is difficult to directly assign the activity of the reporter to the binding of the factor under investigation. Also, this method is low throughput, and there is often bias in the selection of which sites to test. An even more direct approach is to directly delete or mutate sites in vivo and determine whether this alters the expression of the associated target gene. However, the time and cost associated with such an effort make it impractical in most cases.
Thus, it remains unclear whether the many thousands of sites identified by ChIP-seq truly represent functionally active binding events. Continued investigation in this area is required. Specifically, it would be of great value to assess the functionality of descending confidence sites to gain an understanding if at some threshold, or sequencing depth, functional sites are saturated in an experiment.
Chromatin amount
Although, as previously mentioned, the amount of chromatin required for ChIP-seq is far less than for ChIP–chip experiments, obtaining sufficient amounts of chromatin is still a constraint for ChIP-seq. This is in part due to the need for sufficient DNA amounts after size selection, as after this step, linkers are ligated to the extracted DNA and the DNA is amplified by PCR. If an insufficient amount of DNA is present in the PCR, a high level of PCR artifacts will be obtained and sequenced. It is important to note, though, that the amount of starting chromatin required is largely dependent on antibody used. For example, antibodies to histone modifications tend to bind with high affinity, and as little as 200 000–300 000 cells is a feasible starting number for the successful construction of a ChIP-seq library. Antibodies with lower affinities, i.e. for transcription factor antibodies, require substantially more starting material. In our experience, upwards of 30 μg of chromatin can be required, which, particularly for developing tissues, can be an onerous task. However, it is possible to pool isolated nuclei or sonicated chromatin prior to ChIP, or to pool material from successive ChIPs, to achieve this.
Future directions
The transcriptional networks driving endocrine cell development and function are only beginning to be elucidated. In many endocrine tissues, transcription factors critical to normal development and function have been identified, such as PDX1, NEUROG3, etc. in the pancreas (Jensen 2004), NKX2-1, etc. in the thyroid (De Felice & Di Lauro 2004), C/EBPδ, C/EBPβ, and PPARγ, etc. in adipocytes (Cantile et al. 2003), WT1, SF-1, etc. in the adrenal cortex (Hammer et al. 2005), PITX1, PITX2, LHX3, etc, in the pituitary gland (Mullis 2001), as well as many others. Despite the central roles these factors are known to play in the development and function of these organs, only a handful of their direct targets have, in general, been identified. Furthermore, there are over 2000 transcription factors in both the human and mouse genomes, and dozens of endocrine cell types, all with distinct developmental programs. However, as summarized above, to date, ChIP-seq has only been used to identify binding sites for 19 transcription factors from five different cell types, only one of which is a model of an endocrine cell type (Johnson et al. 2007, Robertson et al. 2007, Chen et al. 2008, Marson et al. 2008, Nielsen et al. 2008, Wederell et al. 2008), and no factors have been assayed in any developing tissue. It is therefore clear that ChIP-seq stands to revolutionize our understanding of gene regulation and transcriptional networks in endocrine tissues.
In order to reconstruct transcriptional networks and to elucidate the epigenetic processes in endocrine cell development and function, it is desirable to identify transcription factor binding sites and regions with specific histone modifications in vivo. For such work, ChIP-seq, due to its lower input requirements and ability to sequence deeply, is the most promising technique to apply to endocrine tissues, which, for many biological reasons, are limiting in supply. Indeed, results from applying ChIP-seq in liver tissues (Wederell et al. 2008) indicate that this technique can be applied to other endocrine tissues in future work.
ChIP-seq allows an unbiased perspective into a factor's binding site nucleotide preferences. Currently, transcription factor motifs have predominately been developed from a limited number of related or high-affinity sites, and the generated motifs often underpredict the actual binding potential of the factor. In some cases, in vitro methods such as systematic evolution of ligands by exponential enrichment (SELEX) and DNA-binding arrays have been used to develop motifs (Roulet et al. 2002, Berger & Bulyk 2006). Although these methods are less biased, they do not necessarily reflect the binding of a factor in vivo. Thus, ChIP-based techniques, such as ChIP-seq, are currently the most accurate method to assess the true in vivo binding characteristics of a factor and the application of these techniques to better assess transcription factor binding is likely to be a significant focus of ChIP-seq experiments in future.
Furthermore, ChIP-seq studies have, in part, laid the framework for how histone modifications relate to gene expression. It is clearly of considerable future interest to determine how these modifications are specified and controlled throughout development. These studies have also pointed towards the use of ChIP-seq-derived histone modification patterns for the identification of regulatory regions, allowing the annotation of functional promoter, enhancer, and repressor regions across the genome, even in the absence of prior annotation. As such, the use of ChIP-seq to determine genome-wide histone modification profiles stands to provide a wealth of information on developmental processes, pluripotency, and cancers that are often associated with epigenetic defects (Esteller 2007, Wang et al. 2007, Hirst & Marra 2008). Despite this, to date, ChIP-seq has yet to be used to map the histone modifications occurring genome wide in a developing tissue or cancer model.
One of the greatest powers of data from ChIP-seq experiments lies in the ability to integrate the data with alternative data types. For example, previous studies have compared ChIP-seq data for transcription factors with ChIP-seq data for histone modifications in the same cells (Marson et al. 2008, Robertson et al. 2008). In these cases, the presence of appropriate histone modifications at identified transcription factor binding sites can give higher confidence of a site's functionality. Combining ChIP-seq data for histone modifications with expression data has also proven useful in confirming the association of modifications with gene activity levels (Yu et al. 2008). Associating transcription factor binding with expression data can also produce valuable insights (Johnson et al. 2007), and possibly help identify functional sites. Other similar integrative analyses are likely to be of high value, and this represents one of the most promising directions for ChIP-seq studies in future.
Likewise, ChIP-seq analyses are likely to prove valuable in the context of different comparative analyses, for example comparing transcription factor binding or histone modification changes throughout development, or between cells in different biological states, or between different tissues. Also of interest will be to compare binding sites across species. For transcription factors, this might aid in the identification of functional binding sites, as these are more likely conserved across different species. In spite of this, it is clear that one of the potentially greatest values of ChIP-seq data will be in different integrative and comparative analyses to address specific biological questions, including many that have historically been unapproachable using other techniques.
Conclusions
In the vast majority of endocrine tissues, in vivo binding sites for critical transcription factors are largely unknown. This makes the construction of accurate transcriptional networks impossible. Given this, it is clear that ChIP-seq has the potential to revolutionize our understanding of gene regulation and transcriptional networks. Despite this promise, few studies, to date, have used this technique, and fewer yet have assessed transcription factor binding sites in tissues. It is also clear that, as new high-quality antibodies are developed, as protocols for applying ChIP-seq to smaller amounts of starting material are developed, and as sequencing costs decrease, ChIP-seq will become a more approachable method. To conclude, ChIP-seq is a state-of-the-art technique that surpasses competitive ChIP–chip studies in a number of ways. ChIP-seq studies in endocrine tissues are expected to make invaluable contributions to our understanding of transcriptional regulation and help clarify the transcriptional networks that regulate endocrine cell development and function.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Acknowledgements
The authors would like to acknowledge Gordon Robertson and Pamela Hoodless for their critical review of this manuscript. S J M Jones is a senior scholar of the Michael Smith Foundation for Health Research.
- Received in final form 7 January 2009
- Accepted 9 January 2009
- © 2009 Society for Endocrinology













