- Made available online as an Accepted Preprint 5 December 2007
- Accepted Preprint first posted online on 5 December 2007
Physiological functions of the imprinted Gnas locus and its protein variants Gαs and XLαs in human and mouse
- Physiological Laboratory, School of Biomedical Sciences, University of Liverpool, Crown Street, Liverpool L69 3BX, UK1Laboratory of Developmental Genetics and Imprinting, The Babraham Institute, Cambridge CB22 3AT, UK2Division of Pediatric Endocrinology, Department of Pediatrics, School of Medicine, The John Hopkins University, Baltimore, Maryland 21287, USA
- (Correspondence should be addressed to A Plagge; Email: a.plagge{at}liv.ac.uk)
Abstract
The stimulatory α-subunit of trimeric G-proteins Gαs, which upon ligand binding to seven-transmembrane receptors activates adenylyl cyclases to produce the second messenger cAMP, constitutes one of the archetypal signal transduction molecules that have been studied in much detail. Over the past few years, however, genetic as well as biochemical approaches have led to a range of novel insights into the Gαs encoding guanine nucleotide binding protein, α-stimulating (Gnas) locus, its alternative protein products and its regulation by genomic imprinting, which leads to monoallelic, parental origin-dependent expression of the various transcripts. Here, we summarise the major characteristics of this complex gene locus and describe the physiological roles of Gαs and its ‘extra large’ variant XLαs at post-natal and adult stages as defined by genetic mutations. Opposite and potentially antagonistic functions of the two proteins in the regulation of energy homeostasis and metabolism have been identified in Gnas- and Gnasxl (XLαs)-deficient mice, which are characterised by obesity and leanness respectively. A comparison of findings in mice with symptoms of the corresponding human genetic disease ‘Albright's hereditary osteodystrophy’/‘pseudohypoparathyroidism’ indicates highly conserved functions as well as unresolved phenotypic differences.
The stimulatory G-protein signalling cycle
Heterotrimeric G-proteins that are composed of α, β and γ-subunits, mediate signal transduction from a large number of activated seven-transmembrane receptors to diverse intracellular effector pathways. Many general aspects of G-protein signalling have been covered in recent excellent reviews ( Cabrera-Vera et al. 2003, Wettschureck & Offermanns 2005). The Gs class of α-subunits is characterised by its ability to stimulate adenylyl cyclases (ACs) to produce the second messenger molecule cAMP. It comprises two genes, Gnas (GNAS in human) and guanine nucleotide binding protein, α stimulating, olfactory type (Gnal), which encode Gαs and Gαolf respectively. While Gnas is generally regarded as a ubiquitously expressed gene, Gnal expression is limited to the olfactory epithelium and a few brain regions, in which it largely replaces Gnas expression with very little overlap of the two α-subunits ( Belluscio et al. 1998, Zhuang et al. 2000, Herve et al. 2001). We will focus here on novel findings related to the Gαs-subunit, its gene locus, variant protein isoforms and physiological functions.
G-proteins undergo a cycle of active and inactive states during the signal transduction process as summarised for Gαs in Fig. 1. The inactive form of the G-protein consists of a trimer comprising Gαs in association with β- and γ-subunit complexes at the plasma membrane, whereby Gαs occupies the GDP nucleotide-bound conformation. Membrane anchorage of the α- and γ-subunits is achieved via lipid modifications, in the case of Gαs palmitoylation of the NH2-terminus ( Kleuss & Krause 2003). β- and γ-subunits form a very tight and stable complex ( Wettschureck & Offermanns 2005). A ligand-bound G-protein coupled receptor (GPCR) activates the Gs-protein through promoting the exchange of GDP for GTP on the α-subunit, which results in its dissociation from the receptor and the β- and γ-complexes. The free Gαs subunit can now interact with and stimulate its effector AC until the intrinsic GTPase activity (hydrolysis of GTP) of the α-subunit returns it into the inactive GDP-bound form, which reassociates with the β- and γ-complexes, to enter a new cycle ( Sunahara et al. 1997, Cabrera-Vera et al. 2003). Very little is known about specificities in the interactions between Gαs and the 5 different β-subunits and 12 γ-subunits that have been identified, nor whether specific combinations of these subunits preferentially interact with certain GPCRs.
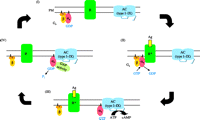
Scheme of the signalling cycle of the trimeric Gs-protein. (I) The inactive, trimeric Gs-protein, consisting of α-, β- and γ-subunits, is associated with the plasma membrane via lipid modifications. The αs-subunit, e.g. Gαs or XLαs, is in its GDP-bound conformation. (II) Agonist binding to a Gs-coupled seven-transmembrane receptor (GPCR) causes a conformational switch in the α-subunit, which also involves an exchange of GDP for GTP, leading to its activation and dissociation from β- and γ-subunits. (III) The active, GTP-bound form of Gαs/XLαs interacts with and activates transmembrane adenylyl cyclases type I–IX, resulting in increased formation of the second messenger cAMP. (IV) The intrinsic GTP hydrolysis activity of Gαs/XLαs, which can be stimulated by GTPase-activating enzymes (GAPs), results in its inactivation and reassociation with β- and γ-subunits.
The Gαs effector AC comprises a family of proteins encoded by nine different genes in mammalian genomes, termed type I–IX, all of which are large transmembrane proteins with a bipartite catalytic domain ( Kamenetsky et al. 2006, Willoughby & Cooper 2007). Although all transmembrane ACs can be stimulated by Gαs, they vary in their responsiveness to additional regulators, e.g. Gαi, G β- and γ-subunits, Ca2+ and protein kinases ( Kamenetsky et al. 2006, Willoughby & Cooper 2007). Most cell types express several AC genes, but certain isoforms dominate in specific tissues ( Hanoune & Defer 2001, Krumins & Gilman 2006, Willoughby & Cooper 2007). In the context of some of the physiological functions of Gαs discussed below, it is noteworthy, for example, that AC III exerts a specific role in brown adipose tissue (BAT). In rodents, AC III expression and AC activity in BAT is transiently increased during the neonatal period, when offspring are especially sensitive to environmental conditions and maintenance of body temperature ( Chaudhry et al. 1996). Stimulation of this signalling pathway results in increased lipolysis and heat production in mitochondria. AC III is strongly upregulated upon stimulation by the sympathetic nervous system, e.g. adrenergic receptor stimulation ( Granneman 1995).
The last step of the G-protein cycle ( Fig. 1), the inactivation of the Gαs subunit and re-association with β- and γ-subunits into the trimeric complex, is triggered by the intrinsic GTPase activity of Gαs ( Cabrera-Vera et al. 2003). Generally, the hydrolysis of GTP by α-subunits is stimulated in vivo by GTPase-activating proteins (GAPs). In the case of Gαs, several proteins have been demonstrated to exert a GAP function, including regulator of G-protein signalling 2 (RGS2; Abramow-Newerly et al. 2006, Roy et al. 2006), AC V itself ( Scholich et al. 1999), RGS-PX1 ( Zheng et al. 2001) and cysteine string protein ( Natochin et al. 2005). Their importance in Gαs signalling in vivo remains to be confirmed.
The Gαs variant XLαs also stimulates cAMP signalling from activated receptors
The identification in PC12 cells of an alternative ‘extra large’ form of the αs subunit, XLαs, brought novel aspects to this signalling pathway ( Kehlenbach et al. 1994). The XLαs protein was found to be mostly identical in sequence to Gαs, apart from the NH2-terminal domain, which was replaced by a different (∼370 amino acid) sequence. As detailed below, the two variants are transcribed from alternative promoters/first exons of the Gnas gene and spliced onto shared downstream exons from exon 2 onwards. The novel, XL-specific NH2-terminus consists of a repeated, alanine-rich motif, a proline-rich domain, a highly charged and cysteine-containing region and a sequence motif that includes a stretch of leucines and is highly conserved among all α-subunits ( Fig. 2A; Kehlenbach et al. 1994). While the repeat motif varies among mammals ( Hayward et al. 1998a, Freson et al. 2003), the other XL-specific domains are well conserved. The function of the proline-rich domain is uncertain; however, the cysteine residues serve for lipid anchorage (palmitoylation) to the plasma membrane similar to Gαs ( Ugur & Jones 2000), while the leucine-containing motif participates in the binding of G-protein β- and γ-subunits ( Kehlenbach et al. 1994, Lambright et al. 1996, Klemke et al. 2000). The ability of XLαs to act as a fully functional Gs-protein, i.e. binding of β- and γ-subunits, activation of AC and coupling of activated receptors, was established in biochemical assays ( Klemke et al. 2000) and in transfections of fibroblasts that lack endogenous Gs-proteins ( Bastepe et al. 2002, Linglart et al. 2006); the characteristics of cAMP signalling were identical for XLαs and Gαs (for rat and human versions) in these transfection studies ( Bastepe et al. 2002, Linglart et al. 2006). Neuroendocrine cell lines that express both proteins endogenously have not yet been analysed (see also Klemke et al. 2000).
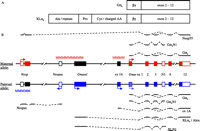
Scheme of the protein domains of Gαs and XLαs encoded by their first exons and of the imprinted Gnas locus. (A) Conserved protein regions encoded by Gnas (Gαs) and Gnasxl (XLαs) first exons. The first exons encode conserved amino acids (βγ) that contribute to the binding of β- and γ-subunits. The Gnasxl specific exon contains further protein regions that are conserved among mammals, e.g. a region with cysteines and charged amino acids (Cys/charged AA) that mediates lipid membrane anchorage, a proline-rich domain (Pro) and a domain containing an alanine-rich repetitive motif. The C-terminus of the two proteins, encoded by exons 2–12 (exons 2–13 in human), is identical. (B) The exon–intron structure (coding exons filled), promoter activities and alternative splicing of the murine imprinted Gnas locus are depicted. The maternally and paternally inherited alleles are indicated in red and blue respectively. Arrows indicate the promoters and transcriptional direction of the individual RNAs. Regions of differential DNA methylation (DMRs) are marked by MMM; DMRs at Nespas/Gnasxl and exon 1A represent imprinting control regions (ICRs). Splicing patterns of the transcripts and encoded proteins are shown above and below the genomic locus. Gnas is expressed biallelically in most tissues, but is silenced on the paternal allele in some cell types (hatched blue box). Gnasxl shows exclusive paternal allele-specific expression and is spliced onto exons 2–12 of Gnas (exons 2–13 in human GNAS). The Gnasxl-specific first exon also contains a second potential open reading frame (ORF) for a protein termed Alex. Nesp is expressed exclusively from the maternal allele. The Nesp55 ORF is contained within the second Nesp-specific exon. Only a single, uninterrupted Nesp-specific exon is found in human. Exon 1A (exon A/B in human) and Nespas produce non-coding, regulatory RNAs; Nespas transcripts exist in multiple spliced and unspliced forms that extend beyond the Nesp exons. Tissue-specific splicing onto exon N1 exclusively in neural tissues leads to premature transcription termination and expression of a truncated XLN1 protein (existence of a corresponding GαsN1 protein is uncertain).
While Gαs is regarded as being more or less ubiquitously expressed, XLαs shows a much restricted expression pattern, being mostly confined to neural and endocrine tissues ( Pasolli et al. 2000, Pasolli & Huttner 2001, Plagge et al. 2004). At embryonic stages, XLαs is already detectable from mid-gestation onwards in regions of neurogenesis and in early differentiating neurons, mainly in areas of the midbrain, hindbrain and spinal cord, including the sympathetic trunk and ganglia ( Pasolli & Huttner 2001). At later embryonic stages expression was also found in the hypothalamus and the pituitary (adenohypophysis and pars intermedia). In the neonatal brain, XLαs expression is confined to distinct regions of the midbrain and hindbrain, e.g. the centre of the noradrenergic system of the brain (locus coeruleus), laterodorsal tegmental nucleus, motor nuclei that innervate orofacial muscles (hypoglossal, motortrigeminal and facial nuclei), as well as scattered cells in the medulla oblongata ( Plagge et al. 2004). Further, sites of expression include the neuroendocrine pituitary (pars anterior and intermedia), the catecholaminergic adrenal medulla and some peripheral tissues, e.g. white adipose tissue (WAT) and BAT, pancreas, heart, kidney and stomach ( Plagge et al. 2004). There are indications that this expression pattern changes towards adulthood, as no XLαs was detected in adult adipose tissues, kidney and heart, but expression persists in brain, pancreatic islets, the pituitary and adrenal glands ( Pasolli et al. 2000, Xie et al. 2006).
The Gnas locus: alternative promoters, splicing and regulation by genomic imprinting
Although the location of the Gαs encoding Gnas gene on mouse distal chromosome 2/human chromosome 20q13.2–q13.3 and its exon–intron structure had been known for some time ( Blatt et al. 1988, Kozasa et al. 1988, Gejman et al. 1991, Levine et al. 1991, Rao et al. 1991, Peters et al. 1994), and despite some early indications for alternative upstream promoters ( Ishikawa et al. 1990, Swaroop et al. 1991), the full complexity of the Gnas locus was only discovered through work in a different field, i.e. genomic imprinting. Imprinting affects a small number of genes in the mammalian genome, currently comprising ∼90 identified transcription units (see databases: http://igc.otago.ac.nz/home.html and http://www.mgu.har.mrc.ac.uk/research/imprinting/index.html). It describes a phenomenon of gene regulation in mammals, whereby one of the two chromosomal alleles is silenced depending on its parental origin. Thus, an imprinted gene is expressed from either the paternally or the maternally inherited chromosome, and this monoallelic, parent of origin-dependent transcription is achieved through mechanisms of DNA methylation, as well as chromatin modifications ( Reik & Walter 2001, Morison et al. 2005, Edwards & Ferguson-Smith 2007).
Separate screens for imprinted genes in human and mouse resulted in the identification of the XLαs-specific first exon of the Gnas locus and an additional exon and promoter, which initiates a transcript that also splices onto downstream Gnas exons but encodes an unrelated, previously identified protein termed Nesp55 ( Fig. 2B; Hayward et al. 1998a, b, Kelsey et al. 1999, Peters et al. 1999). The Gnas locus is now known to comprise a complex arrangement of three protein-coding and two non-coding transcripts regulated by imprinting mechanisms. We will describe the murine locus here, but most features are conserved in humans. As the mechanisms of regulation of the locus by genomic imprinting are currently under much investigation, we will only focus on the main characteristics here, but see Peters et al. (2006) for a recent review.
The protein coding transcripts
The three protein transcripts Gnas, Gnasxl and Nesp each initiate at separate promoters/first exons, but share most of the downstream exons ( Fig. 2B; Plagge & Kelsey 2006, Weinstein et al. 2007). The Gαs encoding Gnas transcript is composed of 12 exons (13 in human, due to an additional intron interrupting exon 9). Most cell types express two variants of the Gαs protein, a small (45 kDa) and a long 52 (kDa) version, which are functionally equivalent ( Graziano et al. 1989, Levis & Bourne 1992) and are generated through alternative splicing of the 15 codons comprising exon 3. Both Gαs versions can vary further by the inclusion of a single serine residue, added through usage of an alternative splice acceptor site at exon 4 ( Bray et al. 1986, Kozasa et al. 1988). The Gnas promoter and exon 1, which encodes amino acids 1–45 of Gαs, do not carry primary marks of genomic imprinting ( Liu et al. 2000b) and in most tissues transcription occurs equally from both alleles. In a subset of tissues or cell types, however, expression is monoallelic and restricted to the maternal allele, e.g. in proximal renal tubules, anterior pituitary, thyroid gland and ovary ( Yu et al. 1998, Hayward et al. 2001, Germain-Lee et al. 2002, 2005, Mantovani et al. 2002, 2004, Liu et al. 2003); this is relevant to human inherited disorders that are associated with hormone resistance symptoms, as discussed below. Imprinting of Gnas in adipose tissue is still contentious, as some studies showed predominant maternal allele-specific expression ( Yu et al. 1998, Williamson et al. 2004), while others found no such preference ( Mantovani et al. 2004, Chen et al. 2005, Germain-Lee et al. 2005). It remains to be clarified whether these discrepant data reflect the analysis of different developmental stages, implying a change in the imprinting status of the Gnas transcript in adipose tissue during the lifetime. In general, tissue-specific imprinting of Gnas has been difficult to demonstrate, since a small amount of transcripts derived from the paternal allele is often detected among the majority that stems from the maternally inherited allele. Whether this is due to incomplete silencing of the paternal allele or a mixture of cell types with imprinted and non-imprinted expression in the tissue samples analysed is unresolved.
A second promoter and first exon are located ∼30 kb upstream of Gnas exon 1 and initiates the Gnasxl transcript ( Fig. 2B; Hayward et al. 1998a, Kelsey et al. 1999, Peters et al. 1999), which is spliced onto exon 2–12 of Gnas. This splice form retains the Gnas open reading frame (ORF) and translates into the XLαs protein as a NH2-terminal variant of Gαs ( Kehlenbach et al. 1994). In contrast to Gnas, the Gnasxl promoter is silenced on the maternal chromosome and activates transcription exclusively from the paternal allele. Apart from the full-length Gnasxl transcript, a prominent truncated form, encoding the protein XLN1, is found in neuroendocrine tissues only (brain, pituitary, adrenal medulla; Klemke et al. 2000, Plagge et al. 2004). This truncation is due to alternative, neural tissue-specific splicing of exon N1, which is located between exons 3 and 4 and contains a termination codon and polyadenylation signal. Originally, exon N1 was described as causing neural-specific truncation of the Gnas transcript ( Crawford et al. 1993) but, in contrast to XLN1 ( Klemke et al. 2000), it remains uncertain whether a corresponding GαsN1 protein is stably expressed. The neural N1 proteins retain the residues for membrane anchorage and part of the domain interacting with β- and γ-subunits ( Klemke et al. 2000), but lack the major functional domains that are encoded by the downstream exons as well as further residues for interaction with β- and γ-complexes ( Lambright et al. 1996). The significance of the exon N1 splice forms, if any, remains to be determined.
The complexity of the Gnasxl transcript is further increased through the highly unusual feature in mammalian mRNAs of a second potential ORF, which is shifted by +1 nucleotide, begins a short distance downstream of the XLαs start codon and terminates at the end of the Gnasxl-specific exon ( Klemke et al. 2001). This ORF encodes a protein termed Alex, which is conserved, but unrelated to G-proteins ( Klemke et al. 2001, Nekrutenko et al. 2005). Although Alex was detected in PC12 cells and human platelets ( Klemke et al. 2001, Freson et al. 2003), its abundance, expression level and significance in vivo remain unclarified.
As a third promoter for a protein-coding transcript within the Gnas locus, the Nesp promoter and first exon are located ∼15 kb upstream of the Gnasxl exon ( Hayward et al. 1998b, Kelsey et al. 1999, Peters et al. 1999). Although the single human NESP-specific exon is interrupted by a short intron in the mouse genome, the downstream splicing onto exons 2–12 of Gnas is conserved and occurs similarly to Gnasxl and Gnas itself. Nesp is imprinted in an opposite way to Gnasxl being expressed only from the maternally derived allele ( Hayward et al. 1998b, Kelsey et al. 1999, Peters et al. 1999). The ORF, which encodes the neuroendocrine secretory protein of Mr 55 000 ( Ischia et al. 1997), is confined to the Nesp-specific exon, and the shared downstream exons function as 3′-untranslated sequence. The Nesp55 protein has similarities with the chromogranin family, is associated with secretory vesicles in neuroendocrine cells and is regarded as a marker for the constitutive secretory pathway ( Fischer-Colbrie et al. 2002). Little is known about its molecular function, but the protein is processed into peptides to variable extent in different cell types ( Lovisetti-Scamihorn et al. 1999). In agreement with its predominant expression in the nervous system and endocrine tissues ( Bauer et al. 1999a, b), mice deficient for Nesp55 show a behavioural phenotype, specifically an altered response to novel environments ( Plagge et al. 2005, Isles et al. manuscript in preparation) but, in contrast to Gαs- and XLαs-deficient mice (see below), they exhibit no major effects on development, growth or metabolism.
Non-coding transcripts and imprinting marks
The complexity of the Gnas locus is not limited to the protein-coding transcripts, but is increased by the occurrence of non-coding transcripts and differentially methylated regions of DNA (DMRs). As noted above, we will only briefly describe how these features relate to how imprinting in the locus is controlled (see also Peters et al. 2006).
Two untranslated transcripts are produced from separate promoters within the locus ( Fig. 2B). The paternal allele-specific exon 1A transcript (exon A/B in human) is initiated ∼2.4 kb upstream of Gnas exon 1 ( Ishikawa et al. 1990, Swaroop et al. 1991, Liu et al. 2000b, Peters et al. 2006) within a CpG dinucleotide-rich cis-regulatory region that is methylated on the maternal allele (exon 1A DMR). This transcript also splices onto exon 2 of Gnas. The second non-coding RNA, Nespas, begins ∼2.1 kb upstream of the Gnasxl-specific exon, but it is transcribed in the opposite direction, i.e. antisense to Nesp ( Hayward & Bonthron 2000, Wroe et al. 2000, Williamson et al. 2006), and is transcribed solely from the paternal allele from within a CpG-rich DMR (methylated on the maternal allele; Coombes et al. 2003). An increasing evidence points towards a role for such non-coding RNA in the regulation of the imprinted, monoallelic expression of the coding transcripts ( Pauler et al. 2007).
The DMRs at exon 1A and Nespas have been shown to be of central importance for the imprinting of the locus ( Williamson et al. 2004, 2006, Liu et al. 2005). At both sites, differential methylation of the maternal allele is established in oocytes and maintained after fertilisation and into adulthood in all somatic tissues ( Liu et al. 2000b, Coombes et al. 2003). Such germline differences in DNA methylation are characteristic of imprinting control regions (ICRs; Spahn & Barlow 2003). A third DMR located at the Nesp promoter is unmethylated in oocytes and sperm, but acquires methylation on the paternal allele during embryonic development ( Liu et al. 2000b, Coombes et al. 2003).
The roles of the exon 1A and Nespas DMRs have been demonstrated through targeted deletion in mice ( Williamson et al. 2004, 2006, Liu et al. 2005). These studies show that the exon 1A region controls the tissue-specific imprinting of Gnas without affecting the upstream transcription units ( Williamson et al. 2004, Liu et al. 2005). Deletion of the exon 1A DMR and promoter on the paternal (normally unmethylated) allele leads to upregulation in cis of the usually silenced expression of Gnas in imprinted tissues. The exact nature of the silencing mechanism exerted by the paternal exon 1A region on Gnas transcription is unknown at present ( Peters et al. 2006).
Deletion of the Nespas promoter, in contrast, affects the imprinting status of all transcripts of the locus ( Williamson et al. 2006), such that the Nespas DMR can be regarded as the principal ICR for the locus. Thus, when Nespas transcription is ablated on the paternal allele, Nesp and Gnas become derepressed, while Gnasxl and the exon 1A transcript are downregulated. Furthermore, the Nesp DMR loses and the exon 1A DMR gains methylation on the paternal allele ( Williamson et al. 2006). The molecular mechanisms through which this ICR controls the imprinted expression of all transcripts of the Gnas locus remain to be elucidated.
Physiological functions of the gene products as revealed by mutations in mice and humans
It has been known for some time that inactivating mutations in the human GNAS gene are associated with the inherited disorder ‘Albright's hereditary osteodystrophy’ (AHO)/‘pseudohypoparathyroidism’ (PHP; Levine et al. 1980, 1983a, Patten et al. 1990, Weinstein et al. 1990, Davies & Hughes 1993). Fuller Albright and his colleagues originally described a disorder characterised by hypocalcaemia, hyperphosphataemia and end organ resistance (in proximal renal tubules) to the main plasma Ca2+ regulator parathyroid hormone (PTH), and therefore named the disease PHP ( Albright et al. 1942). As PTH levels are not reduced, but typically elevated, and since GNAS is biallelically expressed in the calcium-reabsorbing thick ascending limb of the kidney, hypercalciuria does usually not occur in these patients. They also described other specific somatic and developmental abnormalities in these patients and the disorder is now known to include the following additional symptoms: a round face with a ‘short, thickset figure’, early closure of the epiphyses with resultant shortening of one or more metacarpals or metatarsals (brachydactyly), s.c. ectopic ossifications, dental hypoplasia, obesity and cognitive abnormalities of varying degrees from learning disabilities to severe retardation ( Albright et al. 1942, 1952, Weinstein et al. 2001, Levine 2002). Albright and colleagues also noticed patients who showed many of the latter physical features, but had normal calcium, phosphate and PTH levels ( Albright et al. 1952). They termed this combination of symptoms, which was not associated with hormone resistance, ‘pseudopseudohypoparathyroidism’ (PPHP). Both conditions are also referred to as AHO, and identical mutations in GNAS that affect the protein coding sequence can cause AHO with or without hormone resistance. It was Davies & Hughes (1993) who described for the first time the association of the syndromes with the parental origin of the mutation. Thus, paternal inheritance of a GNAS exon mutation results in (AHO-)PPHP, while maternal inheritance is associated with additional resistance to PTH (and other hormones, see below; Levine et al. 1983a) and is now termed ‘PHP type Ia’ (PHP-Ia; Weinstein et al. 2001). Some of the typical features of AHO are shown in Fig. 3A–F and are summarised in Table 1; however, not all features are present in all patients.
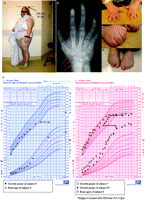
Typical features of AHO. (A) Typical round face and short, obese body habitus (although extreme obesity has been found to be specific for PHP-Ia). (B) X-ray of the hand of an AHO patient showing the striking shortening of the fourth and fifth metacarpals. Arrows are pointing to multiple s.c. ossifications in the hand. (C) Brachydactyly of the hands with marked shortening of the fourth phalanx and metacarpal. (Not the same patient shown in B). The asymmetry in appearance of the hands is common. The arrow points to the very short left thumb referred to as ‘potter's thumb’ or ‘Murder's thumb.’ (D) As a result of the brachymetacarpia, the knuckles are absent and are replaced by dimples when the fist is clenched. This is referred to as ‘Archibald's sign.’ (E) Shortening of the toes is found commonly in AHO. (F) Growth curves of three GH-deficient patients with PHP-Ia (one male (left) and two females (right)) showing the frequent absence of short stature in childhood with resulting short final adult heights. In addition, the pubertal growth spurts are absent. One patient was treated with GH from approximately age 9.5–12 years (referred to as Subject 8) prior to referral. Triangles refer to bone age (no bone age data for Subject 7). Reproduced with permission from Germain-Lee EL, Groman J, Crane JL, Jan de Beur SM & Levine MA 2003 Growth hormone deficiency in pseudohypoparathyroidism type 1a: another manifestation of multihormone resistance. (see comment). Journal of Clinical Endocrinology and Metabolism 88 4059–4069. Copyright 2003, (The Endocrine Society). Signed informed consents were obtained for the patient photographs.
Physiological functions affected by mutations at the GNAS/Gnas imprinted locus in human and mouse
The recent analysis of several mouse models with deficiencies of the individual protein products has deepened our understanding of the associated physiological and endocrine functions ( Plagge & Kelsey 2006, Weinstein et al. 2007). Not surprisingly, homozygous deficiency of Gαs is incompatible with life as embryos die soon after implantation ( Yu et al. 1998, Chen et al. 2005, Germain-Lee et al. 2005). Heterozygous mutations of the different proteins of the Gnas locus cause distinct dysfunctions ( Table 1). In the case of Gαs some aspects of the phenotype vary with the parental origin of the mutation, reflecting its imprinted expression, while other dysfunctions occur after both maternal and paternal transmission, indicating haploinsufficiency of Gαs in some tissues. Heterozygous loss of Gαs in mice recapitulates many aspects of the human disorders, but haploinsufficiency effects seem to be more prevalent in human than in mice. Furthermore, the consequences of loss of XLαs in mice differ and are in several respects opposite to those of specific loss of Gαs, despite their similar capability to activate the cAMP signalling pathway.
Before discussing the physiological and endocrine roles of the different proteins and evaluating the (in some aspects limited) extent of functional conservation between the two species ( Table 1), it should be noted that activating or gain of function mutations of Gnas have also been identified. These are beyond the scope of this review, but have been summarised elsewhere recently ( Hayward et al. 2001, Weinstein et al. 2006, 2007). Furthermore, a separate human disorder associated with the GNAS locus is not due to mutations affecting the protein-coding sequences, but is caused by deregulated imprinting and gene expression control. Originally, it has been characterised by PTH resistance only without clear AHO symptoms and was therefore termed ‘PHP type Ib’ (PHP-Ib; Bastepe & Jüppner 2005). Our current understanding of PHP-Ib is briefly summarised towards the end of this review.
Post-natal physiological functions
All manipulations in mice that lead to lack of maternal allele-specific Gαs or XLαs show an impaired neonatal phenotype with reduced survival ( Cattanach & Kirk 1985, Yu et al. 1998, Cattanach et al. 2000, Plagge et al. 2004, Chen et al. 2005, Germain-Lee et al. 2005).
Heterozygous deficiency of Gαs in mice, generated through deletion of Gnas exon 1, results in a neonatal phenotype on maternal transmission ( Chen et al. 2005, Germain-Lee et al. 2005). The paternally inherited deletion has few consequences at this developmental stage, although some mortality was observed in an inbred strain background ( Germain-Lee et al. 2005). For exon1m−/p+ mice a survival rate to weaning age of 34–51% was observed, again varying with the genetic background used. Most of the losses occur within 3 days after birth, and may result from a severe s.c. oedema, which has been described in several mouse models lacking maternal allele-specific Gαs protein ( Cattanach & Kirk 1985, Yu et al. 1998, Cattanach et al. 2000, Chen et al. 2005). The physiological cause of the oedema, which resolves within a few days after birth, is currently unclear, although a placental dysfunction has been suggested ( Chen et al. 2005, Weinstein et al. 2007). Another consequence of loss of Gαs expression from the maternal allele is the development of profound obesity in adulthood (discussed in detail below). The increase in adiposity arises already during the post-natal stage, as has been documented in mice with maternally inherited mutations of exons 2 and 6 ( Cattanach et al. 2000, Yu et al. 2000, Plagge & Kelsey 2006). Despite their increased lipid accumulation and adipose tissue mass, these mice remain underweight until after weaning.
Comparatively little information on post-natal symptoms is available from case studies of AHO/PHP-Ia patients who carry mutations in GNAS exons on the maternal chromosome. An s.c. oedema has not been documented. However, a few reports describe an early onset of some symptoms characteristic of PHP-Ia at later juvenile or adult stages (see also below; Levine et al. 1985, Weisman et al. 1985, Yokoro et al. 1990, Scott & Hung 1995, Yu et al. 1999, Riepe et al. 2005, Gelfand et al. 2006). From these studies a pattern seems to emerge in which abnormal thyroid function and resistance to thyroid-stimulating hormone (TSH), due to deficient receptor signalling via Gαs, are among the first symptoms detectable: typically, TSH levels are elevated in PHP-Ia at birth ( Levine et al. 1985, Weisman et al. 1985, Yokoro et al. 1990, Yu et al. 1999). The s.c. ossifications can also develop from the first few months onwards, while resistance to PTH, hypocalcaemia and hyperphosphataemia are usually detected only at later stages of infancy or juvenile age ( Eddy et al. 2000, Riepe et al. 2005, Gelfand et al. 2006, 2007). Progressive osseous heteroplasia (POH), a more severe form of extraskeletal ossification with invasion into deeper tissues, can also begin early on, and has been described in association with paternally inherited as well as spontaneously occurring GNAS mutations ( Eddy et al. 2000, Shore et al. 2002, Faust et al. 2003, Gelfand et al. 2007). In general, ossification symptoms are a classical AHO feature, as they can occur upon mutations of the maternal or paternal allele.
Loss of paternally expressed XLαs (through gene targeting of the Gnasxl-specific exon) causes lethality in inbred mouse strains, but 15–20% of mutants survive into adulthood if maintained on an outbred genetic background ( Cattanach et al. 2000, Plagge et al. 2004, Xie et al. 2006). Deficient pups become distinguishable from wild-type littermates within 1 or 2 days after birth, due to a failure to thrive, characterised by severe growth retardation, poor suckling, hypoglycaemia, hypoinsulinaemia, lack of adipose reserves and inertia ( Plagge et al. 2004). This phenotype is most likely related to pleiotropic functions of XLαs in the central nervous system (CNS, e.g. orofacial motornuclei in the context of suckling activity), as well as peripheral tissues that are involved in the maintenance of energy homeostasis (e.g. adipose tissues, pancreas; Plagge et al. 2004). Impairment in neonatal feeding, growth and maintenance of energy balance is found not only in mice with a specific mutation of the Gnasxl exon but also in other mutants that lack XLαs ( Plagge & Kelsey 2006, Weinstein et al. 2007). Thus, mice that carry two copies of the maternally inherited gene locus and no paternal copy (MatDp.dist2) show narrow, flat-sided bodies with reduced adiposity in BAT, hypoactivity, failure to suckle and lethality within a day after birth ( Cattanach & Kirk 1985, Williamson et al. 1998). Two further mutations, a deletion of exon 2 and a point mutation in exon 6 (termed Oed-Sml), affect both Gαs and XLαs upon paternal transmission ( Yu et al. 1998, 2000, Cattanach et al. 2000, Skinner et al. 2002); however, the phenotypes of exon2m+/p− mice and Sml mice are identical in many respects to Gnasxl deficiency ( Yu et al. 1998, 2000, Cattanach et al. 2000, Plagge & Kelsey 2006, Weinstein et al. 2007). The similarity of the phenotypes of these latter two mutations to the Gnasxl mutation indicates that in mice the loss of XLαs is dominant over the simultaneous loss of paternal allele-derived Gαs. Furthermore, as the paternally inherited exon 6 point mutation does not affect the other two proteins expressed from the Gnasxl exon (XLN1 and Alex), this indicates that loss of XLαs is the main cause for the lack of paternal function phenotypes ( Plagge & Kelsey 2006, Weinstein et al. 2007).
The post-natal phenotype of XLαs deficiency improves at around weaning age; no further premature mortality occurs from this stage onwards, although adults remain lean (see below). It is not unlikely that changes in XLαs expression underlie these phenotype changes, since it has been shown for adipose tissue that Gnasxl expression becomes downregulated during the second half of the post-natal period ( Xie et al. 2006).
It is currently uncertain whether XLαs has a similar role in human neonatal physiology. The classical descriptions of patients with AHO/PPHP do not include comparable symptoms. As PPHP patients carry paternally inherited mutations in GNAS-coding exons, similar to exon2m+/p− and Sml mice, XLαs function would be expected to be impaired and dominant over loss of paternally expressed Gαs. However, these mutations cause the same common AHO features as in maternally inherited PHP-Ia (plus additional hormone resistances). A conclusive human case study, which could distinguish between XLαs functions and paternal haploinsufficiency of Gαs by analysing paternally inherited GNAS exon 1 mutations, has not yet been published ( Patten et al. 1990, Fischer et al. 1998, Aldred & Trembath 2000, Mantovani et al. 2000, Long et al. 2007). However, other rare genetic anomalies that disrupt the GNAS locus and XLαs expression, e.g. large chromosomal deletions and maternal uniparental disomies (UPD) of chromosome 20q13.2–q13.3, have been associated with neonatal impairments. Patients with maternal UPD20q13.2–q13.3, who lack a corresponding paternal allele and can be compared with MatDp.dist2 mice described above, show pre- and post-natal growth retardation ( Chudoba et al. 1999, Eggermann et al. 2001, Salafsky et al. 2001, Velissariou et al. 2002). The 20q13.2–q13.3 deletions that include the GNAS locus on the paternal allele, also lead to growth retardation, failure to thrive, feeding difficulties requiring artificial feeding, hypotonia and adipose tissue abnormalities ( Aldred et al. 2002, Genevieve et al. 2005), reminiscent of Gnasxl knockout mice. Although these cases of chromosomal deletions and UPD20s require careful interpretation, as other potentially contributing genes might also be affected, they nevertheless encourage an investigation of PPHP patients for post-natal symptoms, as far as this is feasible and records are available. No null mutations for the Gnasxl-specific exon have been reported in humans, but a polymorphism in the XLαs domain, which results in varied numbers of a 12 amino acid NH2-terminal repeat unit, has been associated with symptoms such as growth retardation, unexplained mental retardation and brachydactyly ( Freson et al. 2001, 2003). Further characterisation of the patients as well as the biochemical functionality of the XLαs repeat variants is required.
Physiological functions in adulthood
The roles of the proteins of the Gnas-locus at adult stages have been characterised in more detail, both in human and mouse ( Table 1). The symptoms common to PHP-Ia and PPHP, which occur independently of parental origin and are due to haploinsufficiency of Gαs in cells with biallelic expression of GNAS, as well as the hormone resistances associated with PHP-Ia upon maternal inheritance of mutations, fully develop towards adulthood. With regard to Gαs, many parallels have now been described between the human diseases and corresponding mouse models, although a role of XLαs in humans remains uncertain.
Hormone resistances
TSH resistance
Mild TSH resistance occurs in most adult patients with PHP-Ia in addition to the usually pronounced PTH resistance described below ( Levine et al. 1983a, Weinstein et al. 2001, Levine 2002). A study using thyroid membranes isolated from a patient with PHP-Ia demonstrated that the defect lies in the signal transduction pathway for TSH, consistent with a defect in Gαs ( Mallet et al. 1982).
Three studies confirmed with strikingly similar results that GNAS is expressed preferentially from the maternal allele in normal human thyroid tissue (mean contribution of the maternal allele: 71.3–75.7%; Germain-Lee et al. 2002, Mantovani et al. 2002, Liu et al. 2003). The fact that the imprinting in the thyroid is partial, e.g. incomplete silencing of the paternal GNAS allele, may provide an explanation for the mild TSH resistance and hypothyroidism found in patients with PHP-Ia. Partial imprinting probably accounts for incomplete hormonal resistance in other tissues as well.
Concurrent studies in Gnas knockout mice with a targeted disruption of exon 1 revealed Gαs imprinting in the thyroid, accompanied by TSH resistance and normal to elevated TSH plasma levels in mice inheriting a disrupted maternal allele, but not in mice with a disrupted paternal allele, similar to humans ( Yu et al. 2000, Chen et al. 2005, Germain-Lee et al. 2005). Although TSH resistance could contribute to other symptoms observed in AHO/PHP-Ia, e.g. short stature and obesity (see below), it seems unlikely that this would be the sole cause in light of the mild degree of hypothyroidism that occurs, as well as the fact that even when patients are successfully treated throughout their lifetime, they are short as adults and also obese ( Long et al. 2007). In addition, mice with maternal Gαs deficiency were obese with normal thyroxine levels, thus implicating other factors in the development of the obesity ( Yu et al. 2000, Chen et al. 2005, Germain-Lee et al. 2005).
PTH resistance
PTH resistance in PHP-Ia patients typically develops over the first several years of life with an elevated PTH usually preceding the hypocalcaemia and hyperphosphataemia ( Werder et al. 1978, Barr et al. 1994, Yu et al. 1999), although there are some patients who do not develop hypocalcaemia until late in adulthood ( Hamilton 1980) and others who maintain normal calcium levels throughout their lifespan ( Balachandar et al. 1975, Drezner & Haussler 1979). Abnormalities in calcium levels most likely result from lack of PTH signalling in the kidney, where it acts on proximal renal tubules as well as distal portions of the nephron. GNAS imprinting and preferential expression from the maternal allele in the kidney occur only in proximal renal tubules, but not in the thick ascending limb or in the collecting ducts, as based on PHP-Ia patients ( Moses et al. 1986, Faull et al. 1991) as well as on results in knockout mouse models ( Yu et al. 1998, Ecelbarger et al. 1999, Weinstein et al. 2000, Germain-Lee et al. 2005). Loss of Gαs from the maternal allele, therefore, disturbs the PTH-mediated inhibition of phosphate reabsorption and its stimulation of 1,25-dihydroxycholecalciferol (activated Vitamin D3) synthesis in the proximal tubules more than the calcium reabsorption in distal parts of the nephron. This combination of effects leads to an imbalance characterised by reduced excretion of phosphate and reduced 1,25-dihydroxycholecalciferol-mediated uptake of calcium via the intestines, as well as reduced mobilisation of calcium from bone, whereas calcium reabsorption in the distal parts of the kidney remains normal and hypercalciuria is rarely observed in PHP-Ia patients ( Weinstein et al. 2000). In some cases of PHP-Ia hypercalcitoninaemia has been reported, which seems to be contradictory at first sight to the indication of hypocalcaemia in these patients ( Wagar et al. 1980, Fujii et al. 1984, Kageyama et al. 1988, Vlaeminck-Guillem et al. 2001, Zwermann et al. 2002). However, resistance to calcitonin signalling (via its Gαs-coupled receptor) and reduced levels of 1,25-dihydroxycholecalciferol, which normally downregulate calcitonin production, have been implicated in causing this symptom ( Vlaeminck-Guillem et al. 2001).
The PTH resistance was also apparent in Gnas knockout mouse models after maternal inheritance of the mutations. On a normal diet, PTH levels were significantly higher (two- to threefold) in m−/p+ mice when compared with wild-type littermates ( Yu et al. 1998, Germain-Lee et al. 2005). On a high phosphate diet, the PTH levels were increased by approximately sixfold over levels in mice fed a standard diet, and the m−/p+ mice showed again significantly elevated levels (2.9-fold) of PTH compared with wild types. The m+/p− mice had PTH levels that were intermediate, trending approximately twofold higher than in wild types, but lower than in m−/p+ mice ( Germain-Lee et al. 2005), indicating that a low level of Gαs expression might normally occur from the paternal allele in renal proximal tubules.
Growth hormone-releasing hormone (GHRH) resistance
GHRH is a hypothalamic hormone, whose receptor on pituitary somatotroph cells is Gs-coupled, leading to stimulation of GH release. It was demonstrated that Gαs is expressed predominantly from the maternal allele in normal pituitary tissue ( Hayward et al. 2001), thereby strengthening the hypothesis that subjects with a defective maternal GNAS allele could have Gαs deficiency in somatotrophs and a reduced GH response to GHRH. Previous scattered case reports of patients with PHP-Ia indicated a broad range of GH status from deficiency to sufficiency ( Urdanivia et al. 1975, Wagar et al. 1980, Faull et al. 1991, Scott & Hung 1995, Marguet et al. 1997). A recent systematic analysis confirmed a markedly increased prevalence of GH deficiency in patients with PHP-Ia due to resistance to GHRH, thus expanding the range of multi-hormone resistances in PHP-Ia ( Germain-Lee et al. 2003, Mantovani et al. 2003). The penetrance of GH deficiency is not 100% though, e.g. ∼68% of PHP-Ia patients ( Germain-Lee et al. 2003, Mantovani et al. 2003), which is in agreement with partial imprinting and incomplete silencing of the paternal allele of GNAS ( Hayward et al. 2001) similar to thyroid and ovary tissues. Structural abnormalities in the pituitary or hypothalamus were not detected, but insulin-like growth factor-I (IGF-I) levels in these patients were subnormal and therefore consistent with GH deficiency. The markedly increased prevalence of GH deficiency has now been confirmed in a much larger group of PHP-Ia patients (Germain-Lee unpublished results); these data argue strongly for the evaluation of GH status in all PHP-Ia patients, since it may be a contributing factor to the other symptoms of short stature and obesity (see below).
Luteinising hormone and follicle-stimulating hormone (LH/FSH) resistance
Patients with PHP-Ia, especially females, usually have evidence of hypogonadism and incomplete sexual maturation ( Namnoum et al. 1998). The features are less noticeable in men, being limited to lack of full pubertal development in some ( Levine 2000). Amenorrhoea or oligomenorrhoea is common ( Wolfsdorf et al. 1978, Levine et al. 1983a, Namnoum et al. 1998), but occasionally there are women with normal menstrual cycles and full-term pregnancies ( Namnoum et al. 1998, Levine 2000). Women show low oestrogen and progesterone levels similar to those in the normal early follicular phase. Elevated LH and FSH levels would be expected in the face of gonadotropin resistance as found in several studies ( Wolfsdorf et al. 1978, Shapiro et al. 1980, Kageyama et al. 1988), but this is not a consistent observation ( Faull et al. 1991, Namnoum et al. 1998). It has been proposed that PHP-Ia patients have a partial sensitivity to gonadotropins that is sufficient for normal follicular development, and also have adequate oestrogen production for appropriate negative feedback, but not enough for normal ovulation. Therefore, resistance to gonadotropins in women with PHP-Ia is more subtle than the other hormonal resistances described above ( Namnoum et al. 1998). This partial gonadotropin resistance is consistent with the majority of GNAS transcripts being derived from the maternal allele in normal ovarian granulosa cells, with a small contribution of transcripts from the paternal allele ( Mantovani et al. 2002).
While it is difficult to assess the true reproductive fitness of PHP-Ia patients ( Namnoum et al. 1998), studies in Gnas exon 1 knockout mice have revealed reduced fertility. Whenever a male or female inherited the disrupted allele from a female (analogous to mother or father having PHP-Ia), the number of progeny born was dramatically decreased ( Germain-Lee et al. 2005). There was no significant effect on the number of offspring born, however, when either parent had inherited a disrupted paternal allele (analogous to mother or father having PPHP).
Common characteristics of PHP-Ia and PPHP
Short stature and brachydactyly
These two somewhat related AHO characteristics are described together in this section, as they are most likely due to common causes. Brachydactyly (brachymetacarpia/brachymetatarsia) are the most reliable signs for diagnosing AHO. The pattern of shortening is usually most notable in the distal phalanx of the thumb and the third through fifth metacarpals ( Fig. 3; Graudal et al. 1988, Levine 2002). Striking bone age advancement also occurs, as described below.
The short stature in PHP-Ia and PPHP most likely results from a combination of multiple factors including GH deficiency, premature bone fusion and absence of a pubertal growth spurt. Of note is that patients are often not short as children ( de Wijn & Steendijk 1982, Germain-Lee et al. 2003, Germain-Lee 2006), but the incidence of short stature in adults with AHO is ∼80% ( Nagant de Deuxchaisnes & Krane 1978). An extensive search of the literature and of historical controls from patients ( Germain-Lee et al. 2003 and unpublished) has revealed that the mean height is ∼5 ft 0.5 in ±0.7 in (153.4 cm ±1.8 cm) in adult males and 4 ft 8.7 in ±0.7 in (144 cm ±1.8 cm) in females.
During childhood PHP-Ia patients with GH deficiency follow the same pattern as other patients with AHO/PPHP, i.e. they are usually not short at this stage ( Fig. 3F). In most GH-deficient PHP-Ia children IGF-I levels were slightly below the normal range, but seemed adequate enough to maintain normal growth velocities. The growth curves of GH-deficient PHP-Ia patients revealed normal stature until approximately early adolescence, at which time there is a cessation in growth and an apparent lack of pubertal growth spurt ( Fig. 3F; Germain-Lee et al. 2003). This is consistent with a premature epiphyseal closure in bones as an important factor causing short stature and brachydactyly in PHP-Ia and PPHP. Both are also characterised by markedly advanced hand-wrist bone ages, thought to be secondary to premature epiphyseal fusion ( Albright et al. 1952, Steinbach & Young 1966, Germain-Lee et al. 2003, Germain-Lee 2006). Several studies have implicated haploinsufficiency of Gαs as being responsible for the premature epiphyseal fusion ( Kobayashi et al. 2002, Bastepe et al. 2004, Tavella et al. 2004, Sakamoto et al. 2005a, b). Biallelic expression of GNAS has been demonstrated in human bone ( Mantovani et al. 2004) and in mouse chondrocytes ( Bastepe et al. 2004). A 50% reduction of Gαs levels in PHP-Ia and PPHP could impair signalling via the PTH/PTH-related peptide receptor, which mediates chondrocyte proliferation and inhibits differentiation. Bone mineral density does not seem to be affected ( Long et al. 2006).
Although GH deficiency cannot fully explain short stature, as both PHP-Ia and PPHP patients have reduced heights, it seems to be playing a supplementary role to that of premature epiphyseal fusion. In support of this notion, adults with PHP-Ia and GH deficiency have a lower height SDS than GH-sufficient PHP-Ia patients ( Germain-Lee et al. 2003). Studies are currently underway to evaluate whether recombinant GH treatment in GH-deficient PHP-Ia children can increase growth velocity and final adult height ( Germain-Lee 2006 and unpublished results). GH treatment could potentially augment linear growth and permit an increased growth velocity prior to the premature fusion of the epiphyses not only in GH-deficient PHP-Ia children, but also in GH-sufficient PHP-Ia and PPHP cases. Also, further comparative investigation of adult patients with PHP-Ia and PPHP is required, to examine the GH status and its influence on short stature in AHO (Germain-Lee et al. unpublished).
The symptom of short stature is reproduced in Gnas knockout mice, as body length of heterozygotes with either a maternally (m−/p+) or a paternally (m+/p−) inherited Gαs mutation is significantly reduced ( Yu et al. 2000, Germain-Lee et al. 2005). Of note is that the m–/p+ females are significantly shorter than their m+/p− counterparts ( Germain-Lee et al. 2005), which raises the possibility that patients with PHP-Ia may be shorter than PPHP patients, due to their additional hormone resistance.
Several further studies using Gnas mouse models have provided evidence that Gαs is important for the control of both the chondrocyte and osteoblast differentiation. In one study, chimeric mice consisting of wild-type and Gαs-deficient cells were generated ( Bastepe et al. 2004). Analysis of the growth plates of chimeric bones revealed that the Gαs-null chondrocytes undergo premature hypertrophic differentiation. This was also detected, although to a lesser extent, in chimaeras with heterozygous mutations ( Bastepe et al. 2004), mimicking the Gαs haploinsufficiency of AHO patients. In a second mouse model, a chondrocyte-specific Gαs knockout, similar premature differentiation of chondrocytes, shortened growth plates, markedly shortened limbs and ectopic cartilage formation were described ( Sakamoto et al. 2005a). In a third study, an osteoblast-specific Gαs knockout, Sakamoto et al. (2005b) described shortened long bones, reduced trabecular and thickened cortical bone and an overall reduced bone turnover. In contrast to the chimaera study, however, heterozygotes with 50% reduced levels of Gαs specifically in chondrocytes or osteoblasts did not show any phenotypic changes. Heterozygous mice with a general Gnas deletion were also reported to be normal with regards to bone length, histomorphology and mineral density (bone volume, osteoblast surface, trabecular thickness, trabecular separation, trabecular number, mineralizing surface and mineral apposition rate; Germain-Lee et al. 2005).
In summary, although Gαs haploinsufficiency causes short adult height and brachydactyly in humans, most likely via ineffective PTH/PTH-related peptide receptor signal transduction resulting in accelerated differentiation of chondrocytes and osteoblasts and premature fusion of the growth plates, clear changes in bone morphology of mice are only observed upon complete loss of Gαs in relevant cells.
S.c. ossifications
S.c. heterotopic ossifications, also known as osteoma cutis, develop in patients with both PHP-Ia and PPHP. AHO is the only monogenic condition, in which de novo ossifications form subcutaneously and remain limited to the skin, causing pain and morbidity for the patients and requiring recurrent surgeries. The aetiology of the ossifications is as yet unknown and is unrelated to abnormalities in serum calcium or phosphorus levels. They can occur spontaneously or in response to minor trauma and are sometimes the presenting sign of AHO ( Izraeli et al. 1992, Prendiville et al. 1992). Patients with GNAS mutations can also develop POH, a more limited disorder, in which severe heterotopic ossifications invade from s.c. tissue into deep connective tissue and skeletal muscle ( Kaplan & Shore 2000, Shore et al. 2002, Gelfand et al. 2007).
Extensive s.c. heterotopic ossifications were found recently in the Gnas exon 1 knockout mouse model of Germain-Lee et al. ( Huso et al. 2007). There are no s.c. ossifications in 3-month-old mice as reported previously ( Germain-Lee et al. 2005); however, because of the increased frequency and size of s.c. ossifications in ageing AHO patients (Germain-Lee unpublished), 12-month-old heterozygous mutants were analysed and revealed extensive heterotopic s.c. bone formation in the dermis ( Huso et al. 2007). Mineral deposits in the areas surrounding hair follicles were detected, and many of these areas contained bone marrow elements, consistent with true s.c. bone formation, which was confirmed by X-ray and computed tomography imaging. There were no differences in the frequency or histology of the s.c. ossifications in mice with either a maternally or paternally inherited mutation, which is analogous to its occurrence in AHO (PHP-Ia and PPHP) patients and consistent with haploinsufficiency/lack of imprinting of Gαs in the relevant cell types ( Levine et al. 1983b, Mantovani et al. 2004).
Cognitive and other CNS abnormalities
AHO is often, but not always, accompanied by cognitive deficits ranging from learning disabilities to severe retardation ( Marguet et al. 1997, Rutter & Smith 1998, Levine et al. 2000, 2002, Weinstein et al. 2001). Reductions in Gαs levels have been associated with cognitive deficiency ( Farfel & Friedman 1986). Patients with medically well-controlled hypocalcaemia and hypothyroidism still present with cognitive deficits, thus excluding these symptoms as potential causes for the neurological findings. Patients with PHP-Ia frequently have seizures, and these may occur before hypocalcaemia is recognised ( Bonadio 1989, Faig et al. 1992). Basal ganglia calcifications can be extensive in PHP-Ia, as they are in regular hypoparathyroidism, and can sometimes lead to movement disorders ( Blin et al. 1991, Dure & Mussell 1998). Abnormalities in olfaction and hearing have also been reported in PHP-Ia and are not present in PPHP ( Henkin 1968, Weinstock et al. 1986, Koch et al. 1990, Doty et al. 1997), suggesting the involvement of GNAS imprinting in the CNS. In addition, abnormalities in taste sensation have been identified in an early study of PHP ( Henkin 1968). In most of the above studies it has not been determined conclusively, however, whether differences occur between PHP-Ia and PPHP, i.e. whether these CNS-related abnormalities are related to imprinting of GNAS or Gαs haploinsufficiency.
Mouse models of Gαs deficiency have provided some evidence for neural functions, although a detailed characterisation is still required ( Yu et al. 1998, Chen et al. 2005). The key question of whether Gnas is imprinted and monoallelically expressed in subregions of the mouse brain remains unclarified for the time being. A first indication that this might be the case was reported in Gnas exon1 knockout mice ( Germain-Lee et al. 2005). Females with a maternally inherited mutation (m−/p+ mothers) neglected their young, resulting in a very high (∼80%) mortality among their pups before weaning. In contrast, females with a paternally inherited mutation (m+/p− females) showed normal mothering behaviour, leading to much less mortality among their offspring (∼27%; Germain-Lee et al. 2005). The poor mothering skills of the m−/p+ females may be reflective of cognitive/sensory defects or hormonal dysfunctions involving the hypothalamus. The behavioural differences between m−/p+ and m+/p− mothers argue against simple haploinsufficiency and in favour of a predominant maternal-allele specific expression of Gnas in some CNS regions.
A role of the maternal allele-derived Nesp55 protein in neural symptoms of AHO/PHP-Ia can be excluded, as mutations in exons 2–13, which often occur in these patients, would only affect the 3′-untranslated sequence of the Nesp transcript without impacting on its coding region. Nevertheless, a mouse knockout of Nesp55 showed a behavioural phenotype, as noted above ( Plagge et al. 2005).
Metabolic deregulation
Obesity is commonly found in AHO subjects and altered metabolic phenotypes are amongst the most interesting effects in Gnas knockout mice. The original knockout in mice revealed an intriguing difference in metabolic phenotype amongst adult mice heterozygous for a disruption of exon 2, depending on parental inheritance. Thus, exon2m−/p+ mice were described as showing accelerated weight gain from around weaning, with increased weights of gonadal white adipose tissue (WAT) and interscapular BAT, whereas exon2m+/p− mice remained underweight with reduced WAT and BAT weights ( Yu et al. 2000). Further examination revealed that exon2m−/p+ mice did not, paradoxically, have increased food intake, but reduced ambulatory activity and resting metabolic rate, whereas exon2m+/p− mice had increased activity and metabolic rate, and a tendency towards hyperphagia.
With more recent, transcript-specific knockouts, the basis for these opposing phenotypes has become clearer. The lean, hypermetabolic phenotype can be attributed to loss of paternally expressed XLαs (or other translation products of the Gnasxl transcript), as it is also present in Gnasxlm+/p− mice, but not in Gnas exon1m+/p− mice, which are deficient only for paternally expressed Gαs ( Chen et al. 2005, Xie et al. 2006). And the obese, hypometabolic phenotype can be put down to loss of Gαs from the maternal allele, as an essentially similar phenotype occurs in Gnas exon1m−/p+ ( Chen et al. 2005). Interestingly, mice heterozygous for the exon 1 disruption on the paternal allele (Gnas exon1m+/p−) have a far milder obesity, without significant effects on metabolic rate.
These observations prompt two conclusions. First, mild obesity reflects haploinsufficiency for Gαs, whilst severe obesity reflects the additional and more profound loss of Gαs function in specific sites caused by its imprinted expression. This leads to the conclusion that Gαs expression is imprinted in hypothalamic or hindbrain nuclei regulating metabolic rate; imprinted expression of Gαs in adipose tissues (see below) appears not to be a factor ( Yu et al. 2000). Second, from a comparison of the Gnas exon1m+/p− and Gnasxlm+/p− phenotypes, the physiological effects of XLαs predominate over those of Gαs expressed from the paternal allele.
The physiological basis of the lean/obese phenotypes is not entirely clear and is likely to be complex, but a primary defect in adipose tissues appears to be ruled out. Maternal monoallelic expression of Gαs in adipose tissues could give rise to resistance to the lipolytic activity of sympathetic innervation or circulating catecholamines, however, as discussed earlier, there is disagreement over whether Gnas is imprinted in adipose tissues. In addition, Gnasxl is abundantly expressed in adipose tissues in neonatal mice, but is strongly downregulated around weaning ( Plagge et al. 2004, Xie et al. 2006), implying that the enhanced metabolic rate in adults is not caused by increased sensitivity intrinsic to the tissue. An explicit test of the sensitivity of adipose tissues in the mutants is the metabolic response to an agonist of the adipose-specific β3-adrenoreceptor: such studies have revealed essentially normal responsiveness in Gnas exon2m−/p+ and Gnasxlm+/p− mice ( Yu et al. 2000, Xie et al. 2006). These results rather suggest a differential effect of maternal Gαs and XLαs on sympathetic activity towards adipose tissues, and support for this proposition comes from the finding of reduced urinary excretion of noradrenalin in exon2m−/p+ and increased excretion in Gnasxlm+/p− mice ( Yu et al. 2000, Xie et al. 2006).
In keeping with their lean phenotype, Gnasxlm+/p− and Gnas exon2m+/p− mice have strongly increased insulin sensitivity, as evidenced by improved glucose tolerance and an exaggerated hypoglycaemic response to injected insulin. Euglycaemic–hyperinsulinaemic clamp studies demonstrated increased glucose uptake into skeletal muscle, WAT and BAT. The mutants also respond to an oral triglyceride load with an increased clearance rate ( Yu et al. 2001, Chen et al. 2004, Xie et al. 2006). Gene expression analysis in Gnasxlm+/p− mice reveals a profile in adipose tissues consistent with increased sympathetic activation and induction of genes associated with triglyceride uptake and hydrolysis, lipid oxidation and the adipogenic pathway ( Xie et al. 2006). In contrast, the paucity of expression changes in skeletal muscle of genes associated with energy metabolism suggests that increased energy dissipation in adipose tissues is the principal cause of the elevated metabolic rate of these mutants.
Glucose homeostasis in mice lacking maternal Gαs is somewhat more confusing: there are differences in phenotypes of Gnas exon1 and Gnas exon2m−/p+ mice, which are unexpected, as both are deficient presumably only in Gαs produced from the maternal allele. Gnas exon1m−/p+ mice have insulin resistance and associated serum abnormalities classically associated with obesity, whereas obese Gnas exon2m−/p+ mice are described as having increased insulin sensitivity, coupled to increased insulin-stimulated glucose uptake into skeletal muscle ( Yu et al. 2001, Chen et al. 2005). Part of the reason for these and other discrepancies between the reports on the various Gnas mutants could be put down to variation in experimental design and environment (i.e. age or gender of experimental groups, husbandry) or genetic background. Studies have used outbred CD1 mice or combinations of inbred strains, which has been done because of the poor viability of the mutants on pure backgrounds. It is also possible that the genetic manipulations themselves may have had unforeseen consequences on the expression of other, relevant transcripts in the locus that could modify sensitive phenotypes such as metabolism.
Until recently, there was no recognition that imprinting of GNAS was relevant to the presentation of obesity in AHO. Obesity is described in both PHP-Ia and PPHP, and irrespective of whether inactivating mutations involve exon 1 (specific for Gαs) or the downstream exons common to all protein-coding transcripts; there was certainly no metabolic phenotype reminiscent of mice lacking XLαs. The demonstration of strikingly opposite effects on metabolism in knockout mice has stimulated a re-evaluation of the clinical data and one recent study has concluded that severe obesity is characteristic of PHP-Ia specifically and not PPHP with the mean BMI z-score (±s.e.m.) in PHP-Ia versus PPHP being 2.31 (±0.18) and 0.65 (±0.31) respectively ( Long et al. 2007). This finding is consistent with Gαs imprinting in a pathway leading to obesity in humans as well as in mice.
PHP-Ib, a disorder due to deregulated imprinting of GNAS
PHP-Ib was initially thought to be a distinct disease entity, because it was presented with isolated PTH resistance without the other endocrine anomalies commonly associated with PHP-Ia or the clinical signs typical of AHO. However, mapping studies in four PHP-Ib kindreds located the disease locus in the 20q region containing GNAS, and also found maternal transmission of disease-associated haplotypes consistent with the presumed imprinting of GNAS ( Jüppner et al. 1998). Although a structural defect in Gαs that selectively affects coupling with the PTH/PTHrP-receptor has been found in one PHP-Ib family ( Wu et al. 2001), the great majority of cases appear to arise from defects in GNAS imprinting, and recent clinical investigations have in fact found mild TSH resistance and even AHO-like symptoms in PHP-Ib patients ( Liu et al. 2003, Mantovani et al. 2007, de Nanclares et al. 2007). The most consistent molecular finding in PHP-Ib is loss of methylation of the exon A/B DMR, which has been detected in the majority of familial cases ( Liu et al. 2000a, Bastepe et al. 2001, Linglart et al. 2007). Studies in mice have shown that the equivalent DMR is required for the tissue-specific imprinting of Gnas ( Williamson et al. 2004, Liu et al. 2005). Although the mechanism of action of the exon A/B DMR is unclear, loss of methylation is predicted to cause silencing of the GNAS promoter on the maternal allele specifically in those tissues in which expression is normally monoallelic, thereby resulting in PTH resistance, without the accompanying symptoms of AHO ( Jüppner et al. 2006). One of the original reports was able to map the genetic defect causing the methylation loss >56 kb upstream of the DMR ( Bastepe et al. 2001), indicating the action of a long-range, cis-acting element. Subsequently, a recurrent 3-kb microdeletion in the neighbouring syntaxin-16 (STX16) gene 220 kb upstream of the DMR was identified in PHP-Ib families ( Bastepe et al. 2003), and has now been documented in over 20 unrelated kindreds ( Linglart et al. 2007, Mantovani et al. 2007). Identification of an overlapping deletion has refined the critical region to 1286 bp containing exon 4 of STX16 ( Linglart et al. 2005). STX16 expression appears not to be imprinted and the mechanism by which these microdeletions result in loss of exon A/B methylation is obscure, particularly as mice engineered to carry a deletion of Stx16 exons 4–6 do not have equivalent methylation abnormalities or develop a PHP-Ib-like phenotype ( Fröhlich et al. 2007). Whilst in most PHP-Ib cases methylation loss is limited to exon A/B, in others there are additional methylation changes across the GNAS locus, and these do not have STX16 deletions ( Bastepe et al. 2001, 2003, Linglart et al. 2007). Instead, two families with loss of methylation of the exon A/B, GNASXL and NESPAS DMRs have been found to have deletions and/or rearrangements spanning the NESP exon ( Bastepe et al. 2005). Again, the mechanism by which these deletions result in failure to establish or maintain methylation of the maternal allele is currently unclear. In contrast to these familial forms, most PHP-Ib cases with more extensive methylation defects present as sporadics with no evidence of STX16 or NESP deletions. In some such cases, unaffected sibs have the same maternal 20q13 haplotype, suggesting the presence of a newly acquired mutation in cis or that the defect is not linked to the 20q13 region ( Linglart et al. 2007). It is interesting to note that a ‘maternal hypomethylation syndrome’ has been described in which affected individuals have loss of methylation at more than one maternal DMR, so that some sporadic PHP-Ib cases may be a manifestation of a more global imprinting defect ( Mackay et al. 2006). An intriguing difference between the various forms of PHP-Ib is that sporadics appear to be more severely affected, while as many as 40% of individuals identified with maternally inherited STX16 deletions are asymptomatic ( Linglart et al. 2007). It is not possible at present to exclude ascertainment bias as the basis for this observation, but it might relate to different molecular events in the establishment of the abnormal methylation patterns or how they impact on the regulation of GNAS imprinting.
Concluding remarks
Since the discovery of the complexity of the Gnas locus and its regulation by genomic imprinting, a number of different mouse models with targeted mutations have greatly contributed to our understanding of the physiological functions of the different protein products. Many parallels between phenotypes in mice and human disease symptoms in AHO/PPHP and AHO/PHP-Ia have become apparent ( Table 1), although some differences are unresolved and might be confirmed as species-specific functions. A role of XLαs in humans remains uncertain. Furthermore, the explanation for the opposite metabolic phenotypes in mice with deficiency of maternally expressed Gαs and paternally expressed XLαs respectively which is likely due to their distinct roles in the CNS regulation of homeostasis, constitutes a major task. A detailed description of the mechanisms of genomic imprinting and regulation of monoallelic expression of this locus are beyond the scope of the review, but progress in this field will be exciting and relevant for the human disorder PHP-Ib, since it is associated with defects in the imprinting mechanisms of GNAS.
Acknowledgements
Work in AP's group is funded by The Royal Society and the Medical Research Council of the UK. Work in GK's group is funded by the UK Biotechnology and Biological Sciences Research Council, Medical Research Council and the European Union. Work in ELG-L's group is funded by the US. Food and Drug Administration Orphan Products Development Grant R01 FD-R-002568, Thrasher Research Foundation Grant 02818-8, the National Institutes of Health/National Center for Research Resources Grant M01RR00052 (to Johns Hopkins University School of Medicine General Clinical Research Center), and The Bosworth Family and Friedman Family Funds. Signed informed consents were obtained for the patient photographs which appear in this publication. All human subjects research referenced as ‘Germain-Lee, unpublished’ was approved by the Internal Review Board of the Joint Committee on Clinical Investigation of the Johns Hopkins University School of Medicine, and informed consent was obtained from all subjects, or parent of each subject, before participation. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
- Received in final form 3 December 2007
- Accepted 5 December 2007
- © 2008 Society for Endocrinology
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










