Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease
- Cardiothoracic Pharmacology, Unit of Critical Care Medicine, Cardiac Medicine, Royal Brompton Hospital, National Heart and Lung Institute, Imperial College, London SW3 6LY, UK
- (Requests for offprints should be addressed to J A Mitchel; Email: j.a.mitchell{at}imperial.ac.uk)
Abstract
Pathogens are sensed by pattern recognition receptors (PRRs), which are germ line-encoded receptors, including transmembrane Toll-like receptors (TLRs) and cytosolic nucleotide oligomerisation domain (NOD) proteins, containing leucine-rich repeats (NLRs). Activation of PRRs by specific pathogen-associated molecular patterns (PAMPs) results in genomic responses in host cells involving activation transcription factors and the induction of genes. There are now at least 10 TLRs in humans and 13 in mice, and 2 NLRs (NOD1 and NOD2). TLR signalling is via interactions with adaptor proteins including MyD88 and toll-receptor associated activator of interferon (TRIF). NOD signalling is via the inflammasome and involves activation of Rip-like interactive clarp kinase (RICK). Bacterial lipopolysaccharide (LPS) from Gram-negative bacteria is the best-studied PAMP and is activated by or ‘sensed’ by TLR4. Lipoteichoic acid (LTA) from Gram-positive bacteria is sensed by TLR2. TLR4 and TLR2 have different signalling cascades, although activation of either results in symptoms of sepsis and shock. This review describes the rapidly expanding field of pathogen-sensing receptors and uses LPS and LTA as examples of how these pathways parallel and diverge from each other. The role of pathogen-sensing pathways in disease is also discussed.
Introduction
In humans and other mammals, innate immune responses provide the first line of defence against invading bacterial pathogens. Pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) from Gram-negative bacteria or lipoteichoic acid (LTA) from Gram-positive bacteria are sensed by macrophages and other immune cells to produce an acute response to the pathogens. Our knowledge of how cells of the immune system sense and respond to a pathogen has increased dramatically within the past few years. Pathogens have specific molecular patterns within their structures, referred to as PAMPs (Fig. 1⇓). Cells of the immune system are activated by, or ‘sensed’ by, pathogens via pathogen-sensing receptors, also known as pattern recognition receptors (PRRs). PRRs are germ line-encoded receptors that sense specific PAMPs and activate responses in cells. These responses are usually genomic involving the induction of new genes via well-classified transduction pathways including NFκB and AP-1. PRRs include transmembrane toll-like receptors (TLRs) and cytosolic nucleotide oligomerisation domain (NOD) proteins containing leucine-rich repeats (NLRs).

Relationship between pathogen-associated molecular patterns (PAMPs), pattern recognition receptors (PRRs) and pathogens. Toll-like receptor (TLR), lipoteichoic acid (LTA), lipopolysaccharide (LPS), double-stranded (ds), single-stranded (ss) RNA, peptidoglycan (PepG).
Bacterial LPS and its effects on biological systems
Bacterial LPS from the cell wall of Gram-negative bacteria has been used in the study of inflammation and the regulation of ‘inducible genes’ for decades. Its efficacy as a model of inflammation means that it is used by scientists in many fields of biomedical research. Indeed, LPS is more often used as a model of ‘inflammation’ than innate immunity. Many of the researchers, who use LPS, rarely gave much attention to how it may be sensed at the surface of the cells being studied. However, a complex interaction between TLR4 and other proteins and factors result in the sensing of LPS by cells. First, we shall consider the effects of LPS in vitro and in vivo, which would explain why it is very useful as a model of inflammation. LPS mimics many inflammatory effects of cytokines, particularly TNFα, interleukin-1β (IL-1β) or IL-6. However, LPS has several advantages over cytokines: it is inexpensive, it is not restricted by species differences and it generally provides a more robust response than individual cytokines.
When administered to cells in vitro, LPS induces a plethora of inflammatory and vasoactive genes, including nitric oxide synthase (NOS)II, cyclo-oxygenase-2, endothelin-1, TNF and other cytokines. When LPS is administered i.v. in vivo, it induces a profound shock (Maclean & Weil 1956). The type of shock induced by LPS is striking. There is an initial drop in blood pressure which begins within 5 min of injection, but resolves within 30–60 min. This is followed after several hours, by a later phase shock characterised by a decline in blood pressure, which is resistant to vasoconstrictors (Szabo et al. 1993). The later phase shock is associated specifically with the induction of NOSII in the vascular smooth muscle component of blood vessels (Szabo et al. 1993, Bishop-Bailey et al. 1997). When LPS is injected locally into the peritoneal cavity (Ajuebor et al. 1999, Elmali et al. 2007), foot pads (Cunha et al. 2000), brain (Marchalant et al. 2007) or inhaled into the lungs (Haddad et al. 2002), an inflammatory response is induced which is generally typified by the activation of macrophages and early recruitment of neutrophils. As explained above, for many years, the signalling pathways involved in the actual sensing of the LPS were unknown. However, it was known that the sensing of LPS was greatly enhanced by the presence of serum, and the serum elements were LPS-binding protein (LBP) and CD14.
LBP and CD14
It was originally believed that LPS activated immune cells through a non-specific mechanism that involved the spontaneous intercalation of lipid A into the mammalian lipid bilayer. However, in the early 1980s, reports emerged suggesting that the biological actions of LPS were facilitated many fold by its binding to endogenous proteins. Tobias et al.(1986) identified LBP (Zweigner et al. 2006). LBP binds to the amphipathic lipid A moiety of LPS (Schumann et al. 1990) and facilitates its presentation and transfer to CD14. In 1990, CD14 was first identified as important to LPS sensing when Wright et al.(1990) demonstrated that it acted as a cell surface ‘receptor’ for the LPS–LBP complex. Initially, and for some years, CD14 was thought to be the actual transducing receptor for the LPS. However, CD14 acts as a manoeuvring protein to guide the complex to TLR4 (see below; Fig. 2⇓).
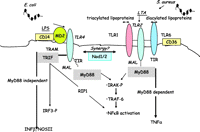
Signalling pathways employed by RLE2 and TLR4. LPS from Gram-negative bacteria, such as Escherichia coli, is manoeuvred to MD2 forming a complex that activates TLR4. The TLR-IL-1 receptor (TIR) domains of TLR4 and TRIF or MyD88 are engaged and signalling occurs via the MyD88 or TRIF pathways leading to NOSII or TNF1 respectively. LTA from Gram-positive bacteria such as Staphylococcus aureus or diacylated lipoproteins are guided by CD36 to activate the TLR2/TLR6 complex. Triacylated lipoproteins activate the TLR2/TLR1 heterodimer. Once activated the TIR domain of TLR2 engages with the TIR domain of MyD88 leading to the induction of ‘MyD88-dependent’ genes including TNFα. Synergy is often seen between TLR and NOD signalling pathways.
Identification and elucidation of the role of TLR4
In the early 1980s, the understanding of TLR-signalling was emerging from fields separate to that of LPS biology. In 1985, a mutant Drosophila that had an unusual appearance was described (Anderson et al. 1985) and was named ‘Toll’, which means ‘weird’. This was caused by a mutation in the Toll gene, which encodes a single-pass transmembrane receptor. In addition to controlling certain developmental processes, toll mediates immunity in flies to fungal and Gram-positive bacteria infections (Gay & Keith 1991, Belvin & Anderson 1996, Lemaitre et al. 1996, Imler & Hoffmann 2001) and was noted to share certain homology with the human IL-1 receptor (Gay & Keith 1991). In the mid-1990s, the field of Toll biology moved beyond flies when several groups identified areas in mammalian gene sequences encoding proteins homologous to toll (Taguchi et al. 1996, Medzhitov et al. 1997, Rock et al. 1998). In another seminal publication, Medzhitov et al.(1997) showed that activation of a human TLR leads to activation of transcription factor NFκB and elements of innate immune responses (Medzhitov et al. 1997).
As mentioned above, LPS is a very active mediator of inflammation in most mammalian systems. However, it has been known for many years that some strains of mice are unresponsive to LPS. For example, LPS is relatively ineffective at inducing responses in the C3H/HeJ or C57BL/10ScCr strains of mouse (Skidmore et al. 1975). Poltorak et al. 1998 showed using positional cloning techniques that mutations of a gene termed the ‘Lps gene’ selectively reduced the ability of C3H/HeJ and C57BL/10ScCr mice to sense LPS. The mutation was shown to correspond to a missense mutation in the third exon of the TLR-4 gene (Poltorak et al. 1998). A similar conclusion was made in 1999 by the Akiras group (Hoshino et al. 1999) who also showed that C3H/HeJ mice have a single point mutation of the amino acid that is conserved among the IL-1/Toll receptor family. They showed, using genetically modified mice in which TLR4 had been deleted, that TLR4 was essential for the sensing of LPS and that its lack of function explained the lack of responsiveness seen in C3H/HeJ mice. Since the publication of these two seminal papers, there has been a large expansion in our understanding of how TLR4 functions in immune responses to LPS, as well as to disease processes without any apparent pathogen link. This may well be due to the fact that TLR4 can not only act as a receptor for LPS, but also for a number of host-derived ligands.
Relationship between CD14, TLR4 and MD2
It is known that MD 2 is required for TLR4 signal transduction. MD 2, like CD14, can be cell bound or secreted and provided to non MD2-bearing cells in serum (Schromm et al. 2001, Fitzgerald et al. 2004). In cells, MD2 is bound to TLR4 (Visintin et al. 2001, Fitzgerald et al. 2004) where it greatly enhances the sensing of LPS (Shimazu et al. 1999). It is believed that MD2 complexes with LPS (provided by CD14) and that the MD2–LPS complex, as apposed to LPS alone, is the ‘true ligand’ for TLR4 (Fitzgerald et al. 2004, Mitchell et al. 2006). However, the precise relationship between TLR4 and MD2 in all instances remains the subject of investigation.
TLRs as a complex family of receptors
By 1998, five human TLR homologues had been identified (Rock et al. 1998). There are now at least 10 TLRs in humans and 13 in mice (West et al. 2006). Both humans and mice have TLRs 1–9. TLR10 is only found in humans, whereas TLR11 is only found in mice. Pathogen ligands (PAMPs) have been identified for TLRs 1–9 (Fig. 1⇑), but the ligands and functions of TLRs 10–13 are less well understood. All of the TLRs contain a TLR-IL-1 receptor (TIR) domain that engages with a TIR domain on one of the number of adaptor proteins. There are two well-defined TLR signalling pathways mediated respectively by MyD88 or TRIF adaptor proteins. MyD88-dependent signalling is common to all of the TLRs, except for TLR3. It involves early phase activation of NFκB via the IL-1R-associated kinase (IRAK) pathway. MyD88-independent signalling is via TRIF and is utilised by TLR4 and TLR3. It is mediated by an interferon regulatory factor 3 pathway, which results in a later phase activation of NFκB. Mal acts as an anchor protein between MyD88 and TIR domains, and may also have a direct role in signalling via interactions with tumour necrosis factor receptor-associated factor 6 (Mansell et al. 2004). Like Mal for MyD88, TRAM seems to link TRIF with the intracellular regions of TLR4. Separate banks of response genes are induced by the MyD88 and TRIF pathways. Importantly, TNFα is a MyD88-dependent gene whereas interferon (IFN) is classified as MyD88 independent, and therefore TRIF dependent (Toshchakov et al. 2002). A list of some of the known ligands for different TLRs is shown in Fig. 1⇑ and the specificities of TLR1, 2, 4 and 6 are discussed in detail below. TLR5 is a cell surface receptor activated by flagellin, a protein expressed on motile bacteria (Gewirtz et al. 2001, Hayashi et al. 2001). TLR3 is an intracellular receptor which is activated by viral double-stranded RNA; TLR7 and TLR8 are activated by single-stranded RNA from viruses. TLR9 is activated by bacterial DNA, which because of its high level of unmethylated CpG dinucleotides is distinct from mammalian DNA (Hemmi et al. 2000). However, it is the sensing of bacterial wall components – LPS (from Gram-negative bacteria) and LTA (from Gram-positive bacteria) – that has been best studied and the differences that have been noted between these PAMPs in many ways define the level of sophistication and utility in the TLR and NOD pathways. As mentioned above, LPS from Gram-negative bacteria is sensed via TLR4. By contrast, LTA from Gram-positive bacteria is sensed by TLR2.
TLR4 versus TLR2
In many respects, LTA and LPS induce similar types of inflammatory responses. Like LPS, LTA will induce shock in laboratory animals (Lonchampt et al. 1992, Kengatharan et al. 1996a,b) and NOSII in macrophages (Kengatharan et al. 1996a,b). However, even if the end pathology may look the same in very broad terms, the signalling and sensing of LPS and LTA could not be more different. The importance of this is clear, when considering identification of new therapeutic targets (see below). As mentioned above, LPS is sensed by TLR4. By contrast, LTA has no affinity for TLR4, but activates TLR2 (Schwandner et al. 1999, Takeuchi et al. 1999a,b; Fig. 2⇑). TLR4 and TLR2 are both cell surface receptors. TLR4 is unique among the TLRs because it recruits both MyD88/Mal and TRIF/TRAM adaptor proteins; TLR2, on the other hand, recruits just MyD88/Mal pathways. The current paradigm would suggest, therefore, that activation of TLR4 allows for a more complex response, in that MyD88 and TRIFF pathways are both activated. However, TLR2 is not thought to act as a monomeric receptor (Triantafilou et al. 2006), but heterodimerises with either TLR1 (Ozinsky et al. 2000) or TLR6 (Takeuchi et al. 1999a,b) to evoke effects. LTA and diacylated fibroblast stimulating lipoprotein-1 (FSL-1) as well as triacylated (Pam3CSK4) lipoproteins all evoke response via TLR2. In early reports, it was thought that TLR2 was the receptor for peptidoglycan (PepG). However, it is known that these observations were due to activation of TLR2 by contaminating lipoproteins in the PepG preparations and not PepG per se (Takeuchi et al. 2002, Travassos et al. 2004). Purified PepG activates NOD receptors.
Gram-positive bacteria contain LTA and lipoproteins. Lipoproteins can be either diacylated or triacylated. The TLR2/TLR6 complex is activated by LTA and diacylated lipoproteins, but not by triacylated lipoproteins (Takeuchi et al. 2001) while the TLR2/TLR1 complex is activated by triacylated lipoproteins, but not by diacylated lipoproteins. It is not yet clear what specific roles TLR2/TLR1 versus TLR2/TLR6 have in immune cells or in disease. Indeed, either complex engages the activation of MyD88-dependent genes and, at this level of signalling there is no clear evidence that the pathways diverge. However, for some ligands (LTA and R-MALP2), activation of the TLR2/TLR6 heterodimer is greatly facilitated by CD36 (Hoebe et al. 2005), which may act similarly to CD14 for TLR4. Interestingly, the level of complexity in pathogen sensing was further illustrated in a recent study by Triantafilou and co-workers showing that while CD36 facilitates TLR2/TLR6 signalling, CD14 enhanced the sensing of ligands by the TLR2/TLR1 complex (Triantafilou et al. 2006).
NOD receptors
In addition to the TLRs, NOD proteins containing NLRs are critically involved in the sensing of bacterial pathogens (Fig. 3⇓). NLRs have been shown to be PRRs for bacterial PepG. There are two well-defined NLRs, NOD1, also known as caspase recruitment domains (CARD4), and NOD2, also known as CARD15. NOD1 senses diaminopimelic acid (DAP)-containing PepG, which is mainly found in PepG of Gram-negative bacteria (Chamaillard et al. 2003), while NOD2 senses the muramyl dipeptide (MDP) present in both Gram-positive and Gram-negative PepG (Inohara et al. 2003). Both NOD1 and NOD2 signal via the adaptor protein RICK (also known as Rip2 or CARDIAK). RICK and NOD1 or NOD2 both contain (CARD) that interact to form a signalling platform comprised of a number of proteins collectively known as the inflammasome. The ensuing signalling cascade leads to activation of caspase-1 and the formation of IL-1β and IL-8. (Inohara et al. 2005, Mariathasan & Monack 2007). Synthetic NOD1 agonists stimulate chemokine production and neutrophil recruitment in vivo (Masumoto et al. 2006), but alone are ineffective in activating monocytic cells in vitro (van Heel et al. 2005). Because of the apparent lack of direct effects on cell signalling induced by activators of NLRs, it is suggested that their role in pathogen sensing is one of co-operation with the TLRs (Takada & Uehara 2006). However, data from our group suggest that the actions of NOD1 vary between cell types and, unlike those seen with LPS, the in vivo effects may be independent of leukocyte activation. Specifically, we have shown that while selective activation of NOD1 in macrophages has no apparent effect, in vascular cells NOD1 activation results in the profound induction of NOSII and shock in vivo (Cartwright et al. 2007). NOD2 is thought to be important in the maintenance of a healthy gut barrier since individuals who carry a defective NOD2 because of a polymorphism have an increased risk of Crohn’s disease (Murillo et al. 2003, Abreu et al. 2005) or other intestinal disorders presumably because the lack of NOD2 compromises gut barrier function to bacteria.
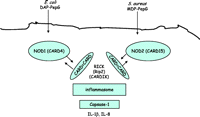
Signalling pathways associated with NOD1 and NOD2. Nucleotide oligomerisation domain (NOD)-1 or NOD2 are cytosolic proteins also known as CARD4 and CARD15 respectively. The CARD domains on NOD1 or NOD2 interact with CARD domains on RICK (also known as Rip2 or CARDIK) to form a signalling platform – the inflammasome that signals via caspase-1 resulting in the induction of IL-1β and IL-8.
PRRs and sensing of whole bacteria
While it is clear that TLR4 is the receptor for LPS and TLR2 for LTA, we should remember that whole bacteria contain a number of different PAMPs each of which activate different PRRs. Gram-negative bacteria have LPS, PepG, DNA and flagellin (for motile forms). Gram-positive bacteria have LTA, lipoproteins, DNA or flagellin. Clearly in the complex setting of a pathogen infection, a number of PRR pathways will be activated. The picture is likely to be even more complex in the case of polymicrobial infections. There is considerable cross talk between TLRs and between the TLR–NOD pathways. In macrophages, ligands for TLR2/1 or TLR2/6 synergise with ligands for TLR4 to induce NOSII or TNFα (Vogel et al. 2003, Paul-Clark et al. 2006). Similarly, co-stimulation of NOD and TLR receptors results in amplification loops in some cells (Akira & Takeda 2004, Strober et al. 2006). Some studies have investigated which of the possible PRRs would predominate in the sensing of whole pathogens.
Viral PRRs
TLR3, TLR7 and TLR8 are intracellular TLRs that are activated by viral PAMPs mediating host immune responses. TLR3 recognises double-stranded RNA (Alexopoulou et al. 2001), while TLR7 (Diebold et al. 2004) and TLR8 (Heil et al. 2004) recognise single-stranded RNA. As mentioned above, TLR3 recruits TRIF only while TLR7 and TLR8 recruits MyD88. Importantly, TLR7 and TLR8 have become important therapeutic targets for the treatment of some specific diseases. Agonists of TLR7, including imiquimod (an imidazoquinoline), cure genital warts caused by human papilloma virus infections (Mitchell et al. 2006). Agonists for TLR7 are successful treatments for neoplastic skin lesion basal cell carcinoma (Stockfleth et al. 2003), metastatic melanoma and other cancers (Mitchell et al. 2006).
Non-pathogenic ligands for TLR receptors
The discovery of TLRs and NLRs has increased our understanding of innate immunity exponentially. However, what is also interesting is the emergence of evidence implicating TLRs and NODs in the pathogenesis of diseases not previously associated with a pathogen element such as atherosclerosis (Bjorkbacka et al. 2004, Michelsen et al. 2004) and asthma (Chaudhuri et al. 2005, Feleszko et al. 2006). Although it is not possible to rule out a role of infection in such disease, it is increasingly recognised that host-derived signals activate TLRs (Mullick et al. 2005). Examples of endogenous ligands of TLRs are heat shock proteins, hyaluronic acid fragments, soluble heparan sulphate, fibrinogen extra domain A, oxidised LDL and β-defensin 2 (Tobias & Curtiss 2005). Work from our own group also suggests that oxidant stress is sensed by TLR2-dependent pathways (Paul-Clarke et al. 2005; http://www.pa2online.org/abstracts/Vol3Issue2.abst159P.pdf). Clearly this observation opens up a central role for TLRs in the wider setting of human disease.
The role of TLRs in the immune-adrenal cross talk
There is an interesting emerging area of research showing a cross talk between TLR and adrenal pathways (Bornstein et al. 2006). It has been known for some time that desensitisation to LPS in vivo is mediated by adrenal hormones (Szabo et al. 1994). TLR2 and TLR4 are expressed in the adrenals and PAMPs such as LPS stimulate glucocorticoid release. Glucocorticoids, in turn, inhibit many actions of LPS in gene induction in cells (Buckingham et al. 2006). TLR2 (Bornstein et al. 2004) and TLR4 (Zacharowski et al. 2006) are directly implicated in the link between bacterial cell wall PAMPs and appropriate adrenal stress response.
Concluding remarks
As we progressively understand more about how different tissues in the body sense pathogens, we anticipate new therapeutic targets will be revealed. This is not only important when we consider diseases caused or exacerbated by pathogens, but also for diseases that are mediated by these pathways via endogenous ligands and with no apparent pathogenic cause.
Acknowledgments
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
- Received 8 February 2007
- Accepted 2 April 2007
- Made available online as an Accepted Preprint 3 April 2007
- Society for Endocrinology










