The leptin-dependent and -independent melanocortin signaling system: regulation of feeding and energy expenditure
- Department of Medicine and Molecular Science, Gunma University Graduate School of Medicine, Maebashi 371-8511, Japan
- 1Department of Regulation Biology, Saitama University, Saitama 378-8570, Japan
- (Requests for offprints should be addressed to M Mori; Email: mmori{at}med.gunma-u.ac.jp)
Abstract
The brain hypothalamus coordinates extra-hypothalamic regions to maintain energy homeostasis through the regulation of food intake and energy expenditure. A number of anorexigenic and orexigenic molecules in the hypothalamic nuclei participate in the control of energy homeostasis. Leptin and pro-opiomelanocortin (POMC)-derived α-melanocyte-stimulating hormone are key anorectic molecules, and the leptin receptor and POMC gene are both expressed in the hypothalamic arcuate nucleus. Although it has been considered that melanocortin signaling is localized downstream to leptin signaling, data have accumulated to support the concept of a leptin-independent melanocortin signaling system. We focus on and review the melanocortin signaling system that functions dependently or independently of leptin signaling in the regulation of energy homeostasis.
Introduction
Obesity forms a fundamental basis of the metabolic syndrome that involves hyperglycemia, hypertension and hyperlipidemia (Wajchenberg 2000, Weiss et al. 2004, Eckel et al. 2005) and is strongly associated with a high incidence of arteriosclerosis and cardiovascular disease (Kenchaiah et al. 2002, Ferreira et al. 2004, Eckel et al. 2005, Saely et al. 2005, Yusuf et al. 2005, Sundstrom et al. 2006). In addition, obesity causes and exacerbates many health problems including non-alcoholic steatohepatitis, sleep apnea, osteoarthritis, cerebrovascular disease, gastroesophageal reflux, gallstones and certain types of cancers (Marceau et al. 1999, Chow et al. 2000, Tilg & Diehl 2000, Stein & Colditz 2004, Angelico et al. 2005, Jacobson et al. 2006). Currently, the incidence of obesity is increasing worldwide. In particular, the rate of obesity involving a body mass index over 30 has been estimated to exceed 30% of the population in the USA (Ogden et al. 2006). The same problem with respect to obesity has occurred in other countries including Europe and Asia (News feature 2004, Gu et al. 2005, Haslam & James 2005, Kawamoto et al. 2005). Analysis of human subjects has shown that both increased adiposity and reduced physical activity are significant and independent predictors of death (Hu et al. 2004, Adams et al. 2006, Jee et al. 2006, Sundstrom et al. 2006).
Increased feeding and decreased energy expenditure lead to obesity (Spiegelman & Flier 2001) and both are regulated essentially by the hypothalamus. The hypothalamus is a known center for the regulation of fundamental behaviors including appetite, thirst, temperature, sexual activity and locomotion. There are complex but integrated interconnections among the hypothalamic nuclei that coordinate the maintenance of energy homeostasis through regulating food intake and energy expenditure (Schwartz et al. 2000, Flier 2004, Schwartz & Porte 2005, Stanley et al. 2005). The individual nuclei of the hypothalamus contain characteristic molecules that exert either an anorexigenic or orexigenic function depending on their hypothalamic locations (Woods et al. 1998, Kalra et al. 1999, Shimizu & Mori 2005). In particular leptin and pro-opiomelanocortin (POMC)-derived α-melanocyte-stimulating hormone (MSH) show strong anorexigenic activities and exert their effects as integrated key molecules in regulating energy homeostasis at the hypothalamic and extra-hypothalamic level (Friedman & Halaas 1998, Schwartz et al. 2000, Stanley et al. 2005).
Although leptin has different effects on the gene expression of hypothalamic anorexigenic molecules such as cocaine- and amphetamine-regulated transcript (CART) and POMC, and on the gene expression of orexigenic molecules such as neuropeptide Y (NPY) and agouti-related peptide (AgRP) (Schwartz et al. 1996, Morrison et al. 2005, Kitamura et al. 2006), in the present review, we discuss what is currently known about the melanocortin signaling system that functions dependently and independently of leptin signaling in the regulation of food intake and energy expenditure.
Brain melanocortin signaling involved in regulating energy homeostasis
POMC is expressed in the skin, the pituitary gland and the hypothalamus (Yeo et al. 2000), and undergoes tissue-specific posttranslational processing (Pritchard et al. 2002). In the anterior pituitary, POMC is processed predominantly to adrenocorticotropic hormone (ACTH) and β-lipotropin (LPH). In the hypothalamus and the neurointermediate lobe of the pituitary, POMC is more extensively processed (Fig. 1⇓). ACTH is further processed to produce α-MSH, N-terminal fragment of POMC is further processed to γ-MSH, and β-LPH is further processed to β-MSH and β-endorphin (Pritchard et al. 2002, Coll et al. 2004). There are species differences in the products of POMC processing; for instance, in humans, γ-LPH is cleaved to produce β-MSH, which is not found in rats (Pritchard et al. 2002, Coll et al. 2004).
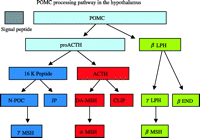
Processing pathway of pro-opiomelanocortin (POMC) in the hypothalamus. N-POC, N-terminal fragment of POMC; JP, junction peptide; DA-MSH, deacetyl MSH; CLIP, corticotropin-like intermediate lobe peptide; LPH, lipotropin; END, endorphin.
The biological effects of POMC-derived fragments are mediated by interaction with specific G protein-coupled seven-transmembrane receptors. Five different receptors have been well characterized. Among these, melanocortin-3 receptor (MC3R) and melanocortin-4 receptor (MC4R) are present in the brain and exert their function in the regulation of energy homeostasis. Whereas α-MSH binds both MC3R and MC4R (MC3/4R), γ-MSH binds MC3R (Millington et al. 2001, Pritchard et al. 2002). Mice deficient in MC3R or MC4R become obese (Huszar et al. 1997, Butler et al. 2000), and mice lacking both MC3R and MC4R have a greater level of obesity compared with mice lacking MC4R only (Chen et al. 2000). Mice deficient in MC3R or MC4R show different characteristics with respect to feeding behavior; MC4R-deficient mice exhibit increased food intake and defects in metabolism, whereas MC3R-deficient mice show increased fat mass without a concomitant increase in food intake even on a high fat diet (Butler et al. 2000, Chen et al. 2000, Sutton et al. 2006). Different responses to peripheral signaling also occur in melanocortin receptor subtypes; MC4R-knockdown mice are resistant to cachexia, but MC3R-deficient mice are highly susceptible to cachexia (Marks et al. 2003). As administration of a specific agonist for MC3R causes a reduction of action potentials in POMC-containing neurons of the arcuate nucleus (Arc) (Cowley et al. 2001), it is reasonable to assume that peripheral administration of an agonist for MC3R would stimulate food intake (Marks et al. 2006). In line with these views, it is proposed that MC3R acts as an inhibitory autoreceptor on POMC neurons. Under MC4R-deficient conditions, the mice no longer respond to the anorectic action of melanotetan II (MT-II), which is a potent agonist not only for MC4R but also for MC3R. Together, these observations lead to the concept that suppression of food intake is primarily due to activation of MC4R (Marsh et al. 1999).
The endogenous molecules antagonizing melanocortin receptors in the brain are agouti and agouti-related peptide (AgRP). Agouti mice (Ay/a) are characterized by ectopic over-expression of agouti protein, which antagonizes skin MC1R and hypothalamic MC3/4R, resulting in altered skin pigmentation and increased food intake (Lu et al. 1994). AgRP, a protein homologous to agouti, is localized in the hypothalamus (Ollmann et al. 1997) and causes increased food intake and body weight (Small et al. 2001); the selective ablation of AgRP-expressing neurons results in acute reduction in feeding (Gropp et al. 2005). However, it is not surprising to find that administration of α-MSH and MT-II causes an apparent inhibitory action on feeding in agouti mice (Fan et al. 1997, Zheng et al. 2002, Martin et al. 2004), because AgRP/agouti also acts as an inverse agonist for a constitutively active MC4R (Haskell-Luevano & Monck 2001, Nijenhuis et al. 2001, Pritchard & White 2005).
There are specific localizations of POMC, MC3R and MC4R gene expression in the brain. Neurons expressing POMC in the brain are restricted to the Arc and the nucleus of the tract solitarius, and POMC gene expression in the Arc is positively regulated by several endogenous molecules including corticotropin-releasing hormone (CRH), insulin, glucocorticoid and leptin (Schwartz et al. 1997, Wardlaw et al. 1998, Kim et al. 1999). The processed products of POMC affect the expression of the POMC gene differently depending on the area of injection; e.g. injection of α-MSH intracerebroventricularly stimulates (Kim et al. 2005) while injection of α-MSH into the paraventricular nucleus (PVN) inhibits expression of the POMC gene (Kim et al. 2002). In contrast to the restricted distribution of the POMC gene, MC3R and MC4R are widely present throughout the brain, and their distributions appear different. MC3R is exceptionally dense in the appetite control-related areas of the hypothalamic nuclei such as the Arc, ventromedial nucleus, and preoptic nucleus (Roselli-Rehfuss et al. 1993, Jegou et al. 2000). On the other hand, MC4R shows intense distribution in the hypothalamic nuclei (Fig. 2⇓) including the PVN, the dorsomedial hypothalamus (DMH) and the lateral hypothalamic area (LHA) (Mountjoy et al. 1994, Kishi et al. 2003, Liu et al. 2003). The important roles that POMC and MC4R play in the regulation of energy balance are evident from the presence of severe obesity in gene-knockout mice (Huszar et al. 1997, Yaswen et al. 1999) and in human subjects with mutations (Krude et al. 1998, Farooqi et al. 2003). Obesity in the knockout mice results from the combined influences of increased food intake and decreased energy expenditure (Huszar et al. 1997, Yaswen et al. 1999, Ste Marie et al. 2000, Balthasar et al. 2005). Individual functions mediated by MC4R occur in different brain regions, and integrated interconnections among the brain regions expressing MC4R have been identified in the regulation of energy homeostasis (Cone 2005). It is of interest to note that in MC4R-deficient mice receiving MC4R-overexpression in the PVN and amygdala, hyperphagia is completely reversed, whereas reduced energy expenditure is unaffected, suggesting that MC4R in the PVN specifically regulates food intake, while MC4R in other regions mainly controls energy expenditure (Balthasar et al. 2005).
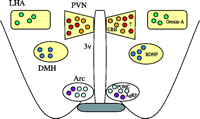
Hypothalamic expression of melanocortin-4 receptor (MC4R) and appetite-control molecules associated with MC4R signaling. MC4R is exceptionally dense (yellow-coloured boxes) in the paraventricular nucleus (PVN), dorsomedial hypothalamus (DMH) and lateral hypothalamic area (LHA). CRH, corticotropin-releasing hormone; BDNF, brain-derived neurotropic factor; POMC, pro-opiomelanocortin; AgRP, agouti-related peptide; Arc, arcuate nucleus; 3v, third ventricle.
Leptin-dependent melanocortin signaling
Leptin originating in peripheral adipose tissue reaches the hypothalamus and binds the active form of the leptin receptor (Ob-Rb) that is localized in the Arc (Friedman & Halaas 1998, Schwartz et al. 2000), where the POMC-expressing neurons co-localize with Ob-Rb (Cheung et al. 1997, Munzberg et al. 2003). Certain physiological conditions that cause changes in the blood level of leptin influence the activity of POMC. For instance, POMC gene expression in the Arc is decreased in leptin-deficient ob/ob mice and in db/db mice with an Ob-Rb mutation (Mizuno et al. 1998). Conversely, administration of leptin causes a significant stimulation of POMC gene expression in the Arc (Schwartz et al. 1996, Morrison et al. 2005, Kitamura et al. 2006), and also increases the frequency of action potentials in Arc POMC neurons (Cowley et al. 2001, van den Top et al. 2004). These data are interpreted as supporting the concept that the melanocortin signaling system basically lies downstream of leptin signaling in the Arc. This concept is compatible with the observations showing the effectiveness of melanocortin signaling in the absence of leptin signaling. Over-expression of POMC ameliorates metabolic abnormalities under the leptin-deficient condition of ob/ob mice (Mizuno et al. 2003), and expression of the POMC gene in the hypothalamic Arc causes reduction of food intake and weight loss in Zucker rats with a leptin receptor mutation (Li et al. 2003).
The hypothalamus also plays a pivotal role in organizing glucose output from the liver (Obici et al. 2002), in which glucose metabolism is mediated by leptin through activation on the central nervous system (Kamohara et al. 1997). In fact, hepatic expression of gluconeogenic enzymes such as glucose-6-phosphate and phosphoenolpyruvate carboxykinase is stimulated by leptin via activation of melanocortin signaling (Gutierrez-Juarez et al. 2004), indicating the downstream localization of melanocortin signaling in relation to leptin signaling.
What molecule(s) in the PVN is(are) involved in the leptin-dependent melanocortin signaling upon the regulation of energy balance (Fig. 2⇑)? Neurons expressing CRH are localized in the PVN and CRH is one of the hypothalamic anorexigenic molecules (Shimizu & Mori 2005). Urocortin is a CRH-related endogenous peptide that induces satiety at the site of the PVN (Currie et al. 2001, Kotz et al. 2002), but urocortin-deficient mice have been observed to show normal basal feeding behavior (Vetter et al. 2002). The CRH-induced satiety function is closely connected with leptin signaling. Not only leptin but also MT-II, an agonist for melanocortin receptors, increase CRH mRNA expression in the PVN (Schwartz et al. 1996, Lu et al. 2003), and the anorexigenic function induced by both leptin and MT-II is hampered by a CRH receptor antagonist (Uehara et al. 1998, Lu et al. 2003). These findings suggest that CRH in the PVN is a downstream mediator of both leptin and melanocortin signaling. However, the anorectic action induced by CRH is not disturbed in MC4R-deficient mice (Marsh et al. 1999) and is not blocked by HSO14, an antagonist for MC4R (Vergoni et al. 1999). These observations are in agreement with the finding that central administration of AgRP does not block CRH-induced feeding suppression (Edwards et al. 2000). Leptin and CRH exert a central action on energy expenditure by activating sympathetic nerve activity (Arase et al. 1988, Luo et al. 2005, Montanaro et al. 2005).
These findings imply that the regulation of energy homeostasis requires the sequential and central relay of leptin and its receptor in the Arc to stimulate melanocortin signaling that is connected with neurons expressing CRH in the PVN (Fig. 3⇓).
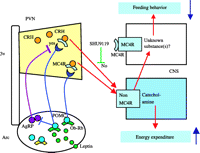
The possible pathway associated with leptin-dependent melanocortin signaling in the regulation of energy expenditure. The CRH-induced energy expenditure is not inhibited by SHU9119. Ob-Rb, leptin receptor; SHU9119, an MC3/4R antagonist; CNS, central nervous system. Other abbreviations are the same as shown in Fig. 2⇑.
Leptin-independent melanocortin signaling
All POMC neurons do not express leptin receptors in the hypothalamus (Cheung et al. 1997, Munzberg et al. 2003), suggesting the existence of a leptin-unrelated melanocortin signaling system.
With respect to energy homeostasis, melanocortin receptors may respond differently to leptin signaling. While leptin administration reduces food intake in MC4R-deficient mice, MC3R-deficient mice do not respond to leptin-induced anorexia, indicating that the ability of leptin to reduce food consumption depends on MC3R, but not on MC4R (Zhang et al. 2005). Previously, it had been shown that neither peripheral nor central administration of leptin induced body weight loss in obese mice deficient in MC4R or in obese agouti mice (Halaas et al. 1997, Marsh et al. 1999). However, these observations seem likely to be the result of leptin resistance found in non-specific obesity (Heymsfield et al. 1999, Ren 2004, Munzberg & Myers 2005), because non-obese MC4R-deficient mice and agouti mice respond to leptin-induced anorexia (Marsh et al. 1999, Martin et al. 2004). These data are compatible, at least in part, with analysis of double-mutant agouti and ob/ob mice that show accelerated obesity and restoration of leptin sensitivity (Boston et al. 1997), indicating leptin-independent roles for melanocortin signaling. In fact, under conditions of leptin resistance such as diet-induced obesity, peripheral administration of MT-II decreases food intake (Pierroz et al. 2002, Bluher et al. 2004) possibly via activation of MC4R (Marsh et al. 1999). These data provide evidence that MC4R-mediated appetite regulation is essentially independent of leptin signaling.
Another example that exhibits leptin-independent melanocortin signaling is the different sympathetic activation by leptin and α-MSH in reducing energy expenditure. α-MSH administration induces a sustained activation of resting energy expenditure (Hoggard et al. 2004) and, conversely, chronic administration of SHU9119, an antagonist for MC3/4R, leads to a decrease in body temperature and physical activity without changing CRH expression in the PVN (Adage et al. 2001). Leptin also induces sympathetic excitation in brown adipose tissue (Luo et al. 2005, Montanaro et al. 2005), but SHU9119 does not block this sympathetic excitation, whereas SHU9119 inhibits sympathetic nerve activity induced by MT-II (Haynes et al. 1999).
A third example of leptin-independent melanocortin signaling is derived from precise comparisons of different metabolic changes between MC4R-knockdown mice and leptin-deficient ob/ob mice. These observations provide insights into the known relationship of the melanocortin system to leptin signaling (Huszar et al. 1997). Transition from a low-fat (12.8%) to a moderately high-fat (25.1%) diet markedly increases body weight gain in MC4R-deficient mice, but not in wild-type or ob/ob mice (Butler et al. 2001). In addition, it has become clear that MC4R signaling, but not leptin signaling, is indispensable for the induction of initial food intake, wheel running activity and oxygen consumption upon exposure to a high-fat diet, suggesting that MC4R takes part in an alternative pathway of the leptin-independent function (Butler et al. 2001).
A fourth example of leptin-independent melanocortin signaling may be the brain fat acid synthase (FAS)-related regulation of energy homeostasis. Alteration of fatty acids in the diet influences their concentration in the hypothalamus (Lam et al. 2005), and fatty acids play pivotal roles not only in the regulation of energy constancy, but also in the modulation of liver glucose metabolism. In the brain, administration of C75 (3-carboxy-4-akyl-2-methylenebutyrolactone) and cerulenin inhibit FAS activity and leads to the accumulation of malonyl CoA, which induces an increase in long-chain fatty acids (LCFAs) and a subsequent decrease in carnitine palmitoyl-transferase-1 (CPT1). Central and peripheral injection of C75 causes a reduction in food intake and body weight (Loftus et al. 2000), and inhibition of hypothalamic CPT1 decreases food intake and glucose production (Obici et al. 2003). Moreover, administration of LCFAs modulates hepatic gluconeogenesis to change the blood glucose level by affecting ATP-sensitive potassium channels in the hypothalamus (Lam et al. 2005, Pocai et al. 2005). Whereas FAS-linked regulation of energy balance is independent of leptin signaling (Loftus et al. 2000), acute administration of C75 prevents the fasting-induced down-regulation of POMC expression and up-regulation of AgRP expression (Shimokawa et al. 2002, Aja et al. 2006). Although C75 has been claimed to be a nonspecific neuronal activator (Takahashi et al. 2004), brain FAS appears likely to require melanocortin signaling in the control of food intake and energy expenditure (Shimokawa et al. 2002, Aja et al. 2006).
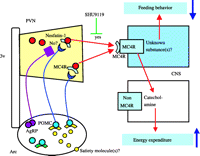
The final example of leptin-independent melanocortin signaling is the hypothalamic role of nesfatin-1 signaling in the regulation of food intake. MC4R is particularly dense in the PVN, DMH and LHA in the hypothalamus (Fig. 2⇑), all areas which participate in the regulation of appetite (Mountjoy et al. 1994, Kishi et al. 2003, Liu et al. 2003). The PVN and DMH are areas implicated in satiety, and the LHA is an area implicated in feeding. In the DMH, one anorexigenic molecule regulated by α-MSH-related MC4R signaling is brain-derived neurotropic factor (Xu et al. 2003), and an MC4R (AgRP)-mediated molecule in the LHA is orexin-A, a potent orexigenic molecule (Zheng et al. 2002). What molecule(s) in the PVN is(are) involved in the leptin-independent melanocortin signaling system? Recently, we identified nesfatin-1 as a novel satiety molecule that is processed from the precursor of NUCB2/nesfatin and distributed in the appetite-controlling hypothalamic nuclei including the Arc and the PVN (Oh-I et al. 2006). Under fasting conditions, nesfatin-1 concentrations are reduced in the PVN. At first, we examined the effect of leptin on gene expression of this molecule. Central administration of leptin does not change gene expression of NUCB2/nesfatin (Fig. 4⇓). In addition, the satiety signal of this molecule is not disturbed in Zucker rats with a leptin receptor mutation, and prior administration of an anti-nesfatin-1 antibody does not block leptin-induced anorexia, suggesting that leptin signaling does not participate primarily in nesfatin-1 signaling. In contrast, central administration of α-MSH markedly stimulates NUCB2/nesfatin gene expression in the PVN (Oh-I et al. 2006). Prior administration of SHU9119 abolishes nesfatin-1-induced feeding suppression, but nesfatin-1 does not show any direct agonistic action on MC3R or MC4R. These observations are interpreted to support the concept that nesfatin-1 signaling involves the leptin-independent melanocortin signaling system in the hypothalamus.

No effects of leptin on NUCB2/nesfatin gene expression in the PVN. Three hours after central administration of vehicle or 20 pmol leptin, the rat brains were analyzed for gene expression using a digitoxigenin-labeled probe of NUCB2/nesfatin cRNA as described previously (Oh-I et al. 2006).
Taken together, the findings imply a sequential and central mechanism in the regulation of energy homeostasis by which an unknown satiety molecule(s) reaches the Arc to stimulate melanocortin signaling-associated neurons expressing nesfatin-1 in the PVN (Fig. 5⇓).
Conclusion
Hypothalamic signaling of leptin and melanocortin plays a key satiety role in regulating food intake and energy expenditure. Obese persons exhibit a consistent increase in blood levels of leptin, but feeding behaviors are not suppressed, implying a conditioned resistance to leptin (Heymsfield et al. 1999, Ren 2004, Munzberg & Myers 2005). Therefore, it is important to evaluate melanocortin signaling that does not depend on leptin signaling in the hypothalamus. The present review raises the possibility that hypothalamic molecules contributing to leptin-independent melanocortin signaling are useful targets for the development of drug therapies to treat human obesity.
Acknowledgments
We thank Ms M Yoshiba and S Adachi for technical assistance. This work was supported in part by grants-in-aid from the Ministry of Health, Labor and Welfare of Japan (to M M). The authors declare that there is no conflict of interest that would prejudice the impartiality of this work.
- Received in final form 7 December 2006
- Accepted 11 December 2006
- Made available online as an Accepted Preprint 27 December 2006
- Society for Endocrinology










