| HOME | HELP | CONTACT US | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
|
|
||||||||
STARLING REVIEW |  |
Endocrinology, Division of Medical Sciences, Institute of Biomedical Research, Medical School, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
(Requests for offprints should be addressed to P M Stewart; p.m.stewart{at}bham.ac.uk)
| Abstract |
|---|
|
|
|---|
Conversely, 11ß-HSD1 has been linked to human obesity and insulin resistance, but also to other diseases in which glucocorticoids have historically been implicated (osteoporosis, glaucoma). Here, the activation of cortisol from cortisone facilitates glucocorticoid hormone action at an autocrine level. The molecular basis for the putative human 11ß-HSD1 ‘knockout’ – ‘cortisone reductase deficiency’ - has recently been described, an observation that also answers a long standing conundrum relating to the set-point of 11ß-HSD1 activity. In each case, these clinical studies have been underpinned by studies in vitro and the manipulation of enzyme expression in vivo using recombinant mouse models.
| Background |
|---|
|
|
|---|
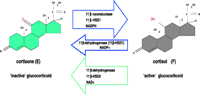
However, studies by Cope and Black showed that the biological activity of any glucocorticoid relates, in part, to the presence of a hydroxyl group at position C11 of the steroid structure, and the inactivation of this group to a C-11 oxo group inactivates the steroid (Cope & Black 1958). Cortisol (Kendall’s compound F), and the principal glucocorticoid in rodents, corticosterone (compound B), are active steroids whereas cortisone and 11-dehydrocorticosterone (compound A), possessing a C11 keto group, are inactive. Therefore, unbeknown to Hench, Kendall and Reichstein, the bioactivity of cortisone was dependent upon conversion to cortisol in the liver. This was supported by studies that showed orally administered cortisone was, in part, excreted as cortisol (Sprague et al. 1950, Axelrod 1953, Burton et al. 1953).
Subsequently, cortisol was characterized as the active ligand and shortly thereafter the first description of tissues converting cortisol to cortisone was published. Amelung and colleagues (1953) discovered that the enzymatic inter-conversion of cortisol in man, and corticosterone in the rodent was performed by the 11ß-HSD enzyme, shown diagrammatically in Fig. 1![]() . 11ß-HSD was designated the number EC1.1.1.146 by the Nomenclature Committee of the International Union of Biochemistry. 11ß-Dehydrogenase activity was subsequently observed in other tissues such as placenta (Osinski 1960, Bernal et al. 1980) and kidney (Jenkins 1966). This was surprising, given that Bush et al.(1968) described a liver enzyme in vivo which was capable of reductase activity, converting cortisone to cortisol. Isotopic studies (Hellman et al. 1971) and clinical studies measuring cortisol/cortisone (F/E) levels in patients with renal disease (Srivastava et al. 1973, Whitworth et al. 1989) confirmed that the kidney was an important site for cortisol to cortisone conversion. Selective venous catheterisation studies indicated significantly lower circulating F/E ratios in renal venous blood compared with peripheral values. In contrast, circulating F/E ratios were much higher in hepatic venous blood, confirming that the liver predominantly converts E to F (Walker et al. 1993). These data suggested the existence of either a single bi-directional enzyme or two distinct isoforms, one catalysing oxidation the other responsible for reduction (Monder & Lakshmi 1989).
. 11ß-HSD was designated the number EC1.1.1.146 by the Nomenclature Committee of the International Union of Biochemistry. 11ß-Dehydrogenase activity was subsequently observed in other tissues such as placenta (Osinski 1960, Bernal et al. 1980) and kidney (Jenkins 1966). This was surprising, given that Bush et al.(1968) described a liver enzyme in vivo which was capable of reductase activity, converting cortisone to cortisol. Isotopic studies (Hellman et al. 1971) and clinical studies measuring cortisol/cortisone (F/E) levels in patients with renal disease (Srivastava et al. 1973, Whitworth et al. 1989) confirmed that the kidney was an important site for cortisol to cortisone conversion. Selective venous catheterisation studies indicated significantly lower circulating F/E ratios in renal venous blood compared with peripheral values. In contrast, circulating F/E ratios were much higher in hepatic venous blood, confirming that the liver predominantly converts E to F (Walker et al. 1993). These data suggested the existence of either a single bi-directional enzyme or two distinct isoforms, one catalysing oxidation the other responsible for reduction (Monder & Lakshmi 1989).
|
|
Although both isozymes are members of the short-chain dehydrogenase superfamily, they share only 21% homology, which is largely at the sites of conserved regions observed in short chain dehydrogenase/reductase (SDR) members. The isozymes are derived from separate gene products (Tannin et al. 1991, Albiston et al. 1994).
| 11ß-Hydroxysteroid dehydrogenase type 2 (11ß-HSD2) |
|---|
|
|
|---|
11ß-HSD2 localisation
The tissue distribution of 11ß-HSD2 has been ascertained by RNA blot hybridisation in human tissues, and the enzyme is expressed in high amounts in adult mineralo-corticoid target tissues (Edwards et al. 1988), such as kidney (Agarwal et al. 1994, Albiston et al. 1994, Stewart et al. 1994) and colon (Whorwood et al. 1994). Immunohistochemistry (Krozowski et al. 1995) and in situ hybridisation studies (Whorwood et al. 1995) localised 11ß-HSD2 in the kidney to the renal collecting ducts in the cortex, most of the medulla, the connecting tubule and the distal convoluted tubule. Expression is also seen in surface epithelial cells in the colon (Whorwood et al. 1994), in salivary epithelial cells (Roland & Funder 1996) and in human skin (Kenouch et al. 1994). The expression of both 11ß-HSD isozymes, 11ß-HSD1 and 11ß-HSD2, in the kidney has been reported and shown to be species-specific, as high levels of 11ß-HSD1 mRNA have been found in the rat kidney, in proximal tubules (Agarwal et al. 1989), while in the human and sheep kidney, 11ß-HSD1 mRNA is barely detectable (Tannin et al. 1991, Yang et al. 1992).
A number of studies demonstrated NAD+-dependent dehydrogenase enzyme activity in human placenta (Brown et al. 1993, Lakshmi et al. 1993) with oxo-reductase predominating in the decidua (Lopez & Craft 1981, Baggia et al. 1990). Studies have shown that 11ß-HSD2 is expressed in the placental trophoblast (Brown et al. 1993, Krozowski et al. 1995, Stewart et al. 1995, Sun et al. 1997). The capacity for 11ß-HSD2 to inactivate glucocorticoids in the placenta is very powerful, and this is supported by the placenta being the most abundant source of the enzyme, per mg of wet weight tissue (Shams et al. 1998). 11ß-HSD2 expression increases to term in the human, baboon and rat placenta (Burton & Waddell 1994, Shams et al. 1998, Pepe et al. 1999). In contrast, in the mouse placenta expression is reduced from gestation day 13 (term=19.5 days) (Brown et al. 1996, Condon et al. 1997).
11ß-HSD2 is also documented in some malignant tissues; as expression is not seen in their non-malignant counterparts this has been implicated as part of the oncogenic process (see later).
The functional role of 11ß-HSD2 in mineralocorticoid receptor (MR)-expressing epithelia is to protect the MR from cortisol. The ligand for the MR in vivo is aldosterone but in vitro studies investigating MR binding (Krozowski & Funder 1983, Arriza et al. 1987) indicated that cortisol, corticosterone, and aldosterone all have equal binding affinities for the MR, with an affinity of around 0.5–1 nM (Arriza et al. 1988). The realisation of the importance of 11ß-HSD2 in this regard came through our understanding of an inherited form of hypertension – ‘apparent mineralocorticoid excess’.
Apparent mineralocorticoid excess (AME)
Defective peripheral conversion of cortisol to cortisone (F-E) in patients with AME, reflecting impaired activity of 11ß-HSD2, was first suggested by Ulick and co-workers in the late 1970s (Ulick et al. 1979).
AME is a monogenic cause of hypertension, with an autosomal recessive inheritance pattern. Approximately 100 cases have been reported worldwide, including several cases of affected siblings (Ulick et al. 1979, Shackleton et al. 1985, Monder et al. 1986, Shackleton & Stewart 1990). AME is characterised by low renin, low aldosterone hypertension, and hypokalaemia. Patients with AME present as children or young adults with severe hypertension and hypokalaemia, suppressed plasma aldosterone concentrations, low plasma renin activity, and extended half-life of plasma cortisol (Ulick et al. 1979, White et al. 1997). The profound hypokalaemia can cause rhabdomyolysis and nephrogenic diabetes insipidus. Other renal abnormalities including renal cysts and nephrocalcinosis have been observed (Milford 1995, Moudgil 2000). Other clinical features include intra-uterine growth restriction (IUGR), short stature, thirst, polyuria and post-natal failure to thrive. The hypertension can be treated with the blockade of mineralocorticoid receptors with the potassium sparing diuretic, Spironolactone, or by a low salt diet, thus it was initially thought that mineralocorticoid excess was causing the disorder. However, all known mineralocorticoids were found to be normal or reduced.
In the 1970s Ulick et al.(1979) suggested that the features of AME could be attributed to defective peripheral conversion of cortisol (F) to cortisone (E) due to impaired activity of 11ß-HSD. Urinary steroid metabolites from AME patients supported this hypothesis as AME was associated with abnormalities in the peripheral metabolism of cortisol, specifically an increase in the urinary excretion of the A-ring reduced cortisol metabolites (tetrahydrocortisol (THF) and allo-THF) versus those of cortisone (tetrahydrocortisone (THE)), suggesting defective 11ß-HSD2 activity (Mune et al. 1995). The excretion of 5ß-cortisol metabolites exceeds that of 5ß-cortisol metabolites, resulting in a high urinary allo-THF/THF ratio and suggesting an additional defect in 5ß-reductase activity (Shackleton et al. 1985, Monder et al. 1986). Typical THF+allo-THF:THE ratios range from 8 to greater than 70 in AME (reference range 0.7–1.1). However, the in vivo assessment of 11ß-HSD2 activity is complicated. The THF+alloTHF:THE measurement provides an index of overall 11ß-HSD activity with the principal contributors being 11ß-HSD1 in the liver and 11ß-HSD2 in the kidney. Therefore, unless the THF+alloTHF:THE is grossly abnormal it is unclear whether 11ß-HSD1 or 11ß-HSD2 is the main contributor to the ratio. The urinary free cortisol/urinary free cortisone ratio (UFF:UFE) may more accurately reflect renal 11ß-HSD2 activity. Normal individuals excrete ~twofold more UFE than UFF (reference range 0.5–0.8), reflecting 11ß-HSD2 activity in the kidney. In AME, UFE excretion is virtually undetectable (Palermo et al. 1996). However, others dispute the suggestion that the UFF:UFE ratio is more sensitive than the THF+alloTHF:THE ratio for detecting decreased 11ß-HSD2 activity (Ferrari et al. 2001).
Lack of 11ß-HSD2 in AME leads to a failure of the ‘protective’ mechanism preventing illicit MR activation and results in cortisol acting as a potent mineralocorticoid (Stewart et al. 1988). However, AME patients do not present with Cushingoid features. In the face of reduced metabolism, the presence of a normal negative feedback system causes a decrease in cortisol secretion rates, thus normal circulating cortisol levels are maintained (Stewart et al. 1988).
Therapeutic treatment of AME
The main aim of treating patients with AME should be to correct life-threatening hypokalaemia and to control blood pressure. Therapeutically, patients have been treated using a wide range of drugs, including potassium supplementation and the MR antagonist Spironolactone. Spironolactone has been of variable benefit, due to high doses being required to block the MR from cortisol activation. Triamterene and Amiloride, potassium sparing diuretics which act by blocking the tubular sodium channels, have also had some success in the treatment of AME. Dexa-methasone has had variable success, and works by suppression of cortisol secretion (dexamethasone itself is not metabolised to any great degree by 11ß-HSD2). Often, additional conventional anti-hypertension treatment is also required, such as angiotensin converting enzyme (ACE) inhibition in order to completely control blood pressure. Interestingly, Captopril (an ACE inhibitor) has been shown to be capable of enhancing renal 11ß-HSD2 activity and it is has been theorised that it could be useful in treating AME patients with partial enzyme activity (Riddle & McDaniel 1994). Finally, a ‘cure’ for AME has been reported in a patient following kidney transplantation presumably because of normal 11ß-HSD2 activity in the transplanted kidney (Palermo et al. 1998).
Molecular basis for AME
At the time of its description, it was thought that mutations in the gene encoding 11ß-HSD1 might be responsible for AME. However, no mutations were identified in the four unrelated AME patients whose gene was first sequenced (White et al. 1994). With the discovery of the 11ß-HSD2 isozyme and the elucidation of its gene structure, the focus for determining the causative mutation for AME moved to 11ß-HSD2. The HSD11B2 gene is 6.2 kb in length containing 5 exons and is located on chromosome 16q22 (Agarwal et al. 1995b). The HSD11B2 gene from AME patients was screened for mutations by sequence analysis, and mutations were initially found to be present in exons 3, 4 and 5 in all the patients studied (Mune et al. 1995). Subsequently, mutations have been identified throughout the gene, although exons 3–4 remain a hotspot for mutations. Mutations on both alleles were found in all cases and thus AME is an autosomal recessive disorder. At present, over 33 different mutations have been defined within HSD11B2 in approximately 60 kindreds; a comprehensive list of the mutations responsible for causing AME is shown in Table 2![]() .
.
|
Recently, nine patients with AME from four families from Oman have been reported. Five mutations, four novel and the previously reported L114Ä6nt (Odermatt et al. 2001) were identified and, for the first time, mutations were identified within exon 1 of HSD11B2 (Quinkler et al. 2004). With the addition of this study, mutations have been identified in all exons and two introns of the HSD11B2 gene.
AME is most commonly found in consanguineous families (Mune et al. 1995, Wilson et al. 1995a,b, Stewart et al. 1996, White et al. 1997). Homozygosity in AME is thought to result from endogamy or a founder effect in Native American families with the R208C and E356Ä1nt mutations, and the L250S, L251P mutation (Mune et al. 1995, Wilson et al. 1995b). The fact that six kindreds are of Native American origin has prompted speculation as to a possible selective advantage of heterozygotes. Such individuals may have an increased ability to conserve salt under conditions of extreme sodium deprivation (White et al. 1997). The Oman population comprises 2.2 million people with a majority (73%) of Omanis with tribal customs consisting of a strong preference for inter-family marriages. In 1995, Wilson et al. (1995a) described a family from Oman with two boys having AME syndrome, the parents and three other siblings being heterozygous for a R208C mutation. The more recent Oman cases derive from four different tribes; however the parents of one kindred and the grandparents of another kindred were second degree relatives. This suggests that the homozygosity of those mutations may be explained by consanguinity in the tribal society. Therefore, the diagnosis of AME should be suspected in patients from tribal societies who present with the features of low birth weight, failure to thrive, polyuria and polydipsia, and hypokalaemia and hypertension (Quinkler et al. 2004). These data suggest that AME may be a common cause of hypertension in certain ethnic groups, such as the Oman population.
AME has generated interest in HSD11B2 as a possible candidate gene for analysis in populations with essential hypertension, more specifically those with low renin, low aldosterone hypertension. Abnormalities in 11ß-HSD2 activity have been shown in patients with essential hypertension either by increases in the plasma [11-3H]cortisol half life or the THF+alloTHF:THE ratio (Walker et al. 1993, Soro et al. 1995). Genetic association studies using polymorphic markers within HSD11B2 have been carried out to determine linkage between 11ß-HSD2 and hypertension. An Italian population was genotyped for a CA repeat polymorphism (11 alleles) in the first intron of HSD11B2, and an association between salt sensitivity (observed in AME hypertension) and shorter CA repeat length was observed, suggesting decreased HSD11B2 expression with shorter CA repeat length. However, functional analysis has failed to support these data (Agarwal et al. 2000). A further study by our group has shown weak associations between the short alleles of the microsatellite within the HSD11B2 gene, and type 1 diabetes mellitus and nephropathy - salt sensitivity and hypertension being phenotypes characteristic of diabetic nephropathy (Lavery et al. 2002). However, linkage and/or association studies to date have mainly been negative, probably because of underpowered studies; a direct genetic link remains to be defined (Brand et al. 1998, Smolenicka et al. 1998).
11ß-HSD2 knockout (KO) mouse
To further assess the role of 11ß-HSD2 in vivo, mice have been generated with targeted disruption of the 11ß-HSD2 gene. The homozygous mutation was generated by removing the genomic fragment encompassing exons 2–5 through specific recombination in mouse embryonic stem cells (Kotelevtsev et al. 1999). The mice appear normal at birth, but around 50% show motor weakness and die within 48 hours of birth. Female and male survivors were fertile and exhibited severe hypertension, hypokalaemia and polyuria, thus exhibiting a typical human AME syndrome phenotype. The epithelium of the distal tubule of the nephron displayed hypertrophy and hyperplasia. Interestingly, the heterozygote mice appeared to be like the wild-type littermates for all parameters studied, and to date no phenotype has been described. The 11ß-HSD2 KO mice will be a useful model to evaluate the role of cortisol in hypertension.
Malignancy and tumorigenesis
It has been well documented that glucocorticoids have an antiproliferative action by inducing cell cycle arrest at the G1 phase (Sanchez et al. 1993, Rogatsky et al. 1997). 11ß-HSD2 expression has now been described in a number of tumours including adrenal adenomas and carcinomas (Coulter et al. 1999), lung adenocarcinomas (Suzuki et al. 2000) and adrenocorticotrophin (ACTH)-producing small cell lung cancer (Parks et al. 1998). Recent studies of bone and the pituitary have shown that both 11ß-HSD isozymes may be expressed at these sites. Specifically, our group has described a shift from predominant 11ß-HSD1 expression in normal tissue to 11ß-HSD2 in tumours (Bland et al. 1999, Cooper et al. 2000, Eyre et al. 2001, Korbonits et al. 2001). In each case the presence of 11ß-HSD2 in neoplastic cells was associated with GR rather than MR expression, suggesting an alternative function for the isozyme that is distinct from its classical role in MR-rich tissues, such as the colon or kidney. In view of the fundamental transregulatory role of the GR and its potential impact on cell proliferation and differentiation, we have hypothesised that abnormal expression of 11ß-HSD2 in GR-rich tissues will confer growth advantage and may be an important component of tumour initiation and/or progression.
| 11ß-Hydroxysteroid dehydrogenase type 1 (11ß-HSD1) |
|---|
|
|
|---|
11ß-HSD1 is a low affinity NADP(H)-dependent bi-directional enzyme, capable of carrying out both 11-oxo-reductase and dehydrogenase reactions, interconvert-ing inactive cortisone and active cortisol (Fig. 2![]() ). The enzyme is microsomal (Ozols 1995) and NADP dependent; in a cell-free system it behaves mainly as a dehydrogenase and no reductase activity is detected in the purified preparation, as described by Ozols (1995). Homogeneous enzyme gave rectilinear Eadie plots and Km constants of 1.83 ± 0.06 µM for corticosterone and 17.3 ± 2.24 µM for cortisol. First order rate constants were an order of magnitude higher for corticosterone than cortisol, but maximal velocities were similar (Lakshmi & Monder 1988).
). The enzyme is microsomal (Ozols 1995) and NADP dependent; in a cell-free system it behaves mainly as a dehydrogenase and no reductase activity is detected in the purified preparation, as described by Ozols (1995). Homogeneous enzyme gave rectilinear Eadie plots and Km constants of 1.83 ± 0.06 µM for corticosterone and 17.3 ± 2.24 µM for cortisol. First order rate constants were an order of magnitude higher for corticosterone than cortisol, but maximal velocities were similar (Lakshmi & Monder 1988).
|
Set-point of 11ß-HSD1 activity
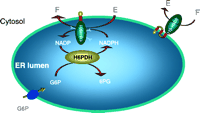
In original purification studies, liver 11ß-HSD1 was shown to be bi-directional although, in contrast with its dehydrogenase activity, the reductase activity was unstable in vitro (Lakshmi & Monder 1988). More recently, a series of studies suggests that the enzyme prefers the reductase direction unless cells are disrupted. This applies in primary cultures of cells from liver (Jamieson et al. 1995), adipose tissue (Bujalska et al. 1997), lung (Hundertmark et al. 1995), hippocampus (Rajan et al. 1996), and vascular smooth muscle (Brem et al. 1995). In a few studies, for example in Leydig cells, 11ß-dehydrogenase activity has been reported in apparently intact cell preparations (Phillips et al. 1989), but others have found predominant 11ß-reduction (Leckie et al. 1998) and argued that some 11ß-HSD1 must be liberated from damaged cells to detect 11ß-dehydrogenase activity. This striking change in directionality between intact cells and homogenates has never been satisfactorily explained, but is likely to reflect the specific intracellular localization of 11ß-HSD1 within the lumen of the endoplasmic reticulum (ER), where neighbouring enzymes may be powerful generators of the reduced co-substrate NADPH. Hexose-6-phosphate dehydrogenase (H6 PDH) is the first enzyme in the microsomal version of the cytosolic based pentose phosphate pathway, and is related to glucose-6-phosphate dehydrogenase (G6 PDH). It generates NADPH through the oxidation of various hexose-6-phosphates including glucose-6-phosphate and appears to be responsible for NADPH provision for 11ß-HSD1 thus regulating its set-point of activity (see later).
The human gene encoding 11ß-HSD1 (HSD11B1)
The human HSD11B1 gene was first cloned and isolated by Tannin et al. in 1991. A rat 11ß-HSD cDNA was used to probe a human cDNA library (testis) and the corresponding human 11ß-HSD cDNA was used to determine chromosomal localisation. HSD11B1 was localised to chromosome 1 by human/hamster cell hybrid panels, and clones containing the HSD11B1 gene were isolated from a chromosome 1 genomic library. The human cDNA predicted a protein of 292 amino acids and was 77% identical at the amino acid level to the rat enzyme (Tannin et al. 1991). The human HSD11B1 gene is localised to chromosome 1 (1q32–41) and consists of 6 exons (Fig. 3![]() ). When first cloned, the gene was thought to be approximately 9 kb in size; however, subsequent studies revealed a much larger than previously recognised intron 4 of approximately 25 kb, expanding the size of the 11ß-HSD type 1 gene to approximately 30 kb.
). When first cloned, the gene was thought to be approximately 9 kb in size; however, subsequent studies revealed a much larger than previously recognised intron 4 of approximately 25 kb, expanding the size of the 11ß-HSD type 1 gene to approximately 30 kb.
|
Structural analysis of 11ß-HSD1
Little is known about the 11ß-HSD1 protein as its crystal structure has yet to be determined. The structure has been modelled on other SDR family members where the crystal structure has been identified.
The amino acid sequence of 11ß-HSD1 was determined by Edman degradation of 11ß-HSD1 purified from rabbit liver microsomes. The protein was determined to have 291 amino acid residues. This sequence was later shown to have 80% homology with human liver 11ß-HSD1. Residues 17–49 were shown to be highly conserved, whereas the C-terminus shows most variability across species (Ozols 1995). The topology of 11ß-HSD1 was elucidated by utilising 11ß-HSD1 constructs with attached FLAG epitopes at the N- and C-terminal regions. 11ß-HSD1 is intrinsic to the ER membrane, the N-terminus region of 11ß-HSD1 traverses the ER membrane and the bulk of 11ß-HSD1 is orientated in the lumen, whereas the N-terminus is cytoplasmic (Odermatt et al. 1999).
Co-factor binding sites for pyridine nucleotides, NADP/NAD were localised to residues 41–47 within the 11ß-HSD1 sequence (GXXXGXG). A putative active site region identified by Tyr-X-X-X-Lys sequence was identified at residue 182 of 11ß-HSD1 (Ozols 1995). This sequence has been implicated as part of the enzyme active site in some members of the short-chain dehydrogenase family (Persson et al. 1991). Mutagenesis studies of Tyr-179 and Lys-183 have shown these residues to be essential for enzymatic activity of 11ß-HSD1 (White et al. 1994). Mutation of the serine residues within the YSASK region of rat 11ß-HSD1 has indicated that these residues play an important role in determining the rate of the catalytic reaction (Obeyesekere et al. 1998).
Within the primary structure of 11ß-HSD1, the protein was found to be associated with oligosaccharides, Asn-X-Ser glycosylation motifs, at three specific sites. The Asn-X-Ser sites are at positions 123–125, 162–164, and 207–209 of the protein. The importance of N-linked glycosylation to 11ß-HSD1 activity has been controversially discussed in the literature. Ozols reported that de-glycosylation of rabbit 11ß-HSD1 resulted in no alteration in oxo-reducing ability (Ozols 1995). However, Agarwal et al.(1990) showed that glycosylation of rat 11ß-HSD1 was essential for dehydrogenase activity (<50%), but did not alter reductase activity. Also muta-genesis of the three potential N-glycosylation sites resulted in either reduced or complete abolition of enzyme activity (Agarwal et al. 1995a). This study was one of the first to show that HSDs could be regulated by post-translational modification. However, the same group also reported that the activity of de-glycosylated 11ß-HSD1 purified from human liver showed no difference to native enzyme activity in both dehydrogenase and reductase directions (Odermatt et al. 1999, Blum et al. 2000a). In support of this finding, recent data conclusively show fully active non-glycosylated 11ß-HSD1 enzyme activity generated in E. coli, with kinetic properties for both dehydrogenase and reductase activities similar to those reported in mammalian systems (Walker et al. 2001). These data suggest glycosylation is not required for correct protein folding or enzyme activity (Blum et al. 2000b). Glycosylation of 11ß-HSD1 may still play a role in preventing protein aggregation, in addition to stabilising the overall structure within the ER (Ozols 1995).
Studies of purified protein have helped to elucidate some sites of post-translational modification, and the enzyme’s orientation within the ER. 11ß-HSD1 has been difficult to purify as the enzyme has remained largely insoluble due to its glycosylation state and N-terminal region. Expression of 11ß-HSD1 constructs in E. coli, containing an N-terminal 6 xHis tag, has yielded a purified (157-fold) protein exhibiting full enzyme activity, with Kms equivalent to those seen in mammalian systems (Walker et al. 2001).
11ß-HSD1 knockout mouse
An important advance in understanding the role of 11ß-HSD1 in glucocorticoid action was the creation of the 11 ß-HSD1 knockout mouse (–/–) (Kotelevtsev et al. 1997). The HSD11B1 homozygous mutant was generated by removing the genomic fragment encompassing exons 3 to 4 through specific recombination in mouse embryonic stem cells.
The –/– mouse is incapable of converting 11-dehydrocorticosterone to active corticosterone - this con-firmed the fact that the only enzyme capable of 11ß-reduction in vivo, at least in mice, is 11ß-HSD1, refuting suggestions of a third isozyme. The mice showed no visible abnormalities, and retained a fully active 11ß-HSD2 isozyme. The mice were fertile, producing normal litters. The adrenal glands of male 11ß-HSD1 –/– mice are enlarged, suggesting adrenocortical hyperplasia. This could be explained by stimulation of the hypothalamic–pituitary–adrenal (HPA) axis to restore corticosterone levels (Holmes et al. 2001). The overcompensation of the HPA axis, leading to greater corticosterone levels than wild-type mice, may be explained by the absence of 11ß-HSD1 in the CNS, removing a level of regulation (Holmes et al. 2001).
The mouse studies highlighted the importance of hepatic 11ß-HSD1 in activating gluconeogenic enzymes and regulating hepatic glucose output. A high fat diet led to 11ß-HSD1 –/– mice that had significantly lower fasting plasma glucose levels than weight-matched litter mates. Glucocorticoids stimulate gluconeogenesis, and phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6 Pase) catalyse key steps of gluconeogenesis. Upon starvation these enzymes are induced by glucocorticoids; however, the 11ß-HSD1 –/– mice failed to show normal induction of G6 Pase and PEPCK. Fed mice accumulated liver glycogen, reflecting lack of glucocorticoid-induced glycogenolysis. Thus 11ß-HSD1 –/– mice are able to resist the hyper-glycaemia seen in obese wild-type mice (Kotelevtsev et al. 1997).
| 11ß-HSD1, human obesity, insulin resistance and metabolic syndrome |
|---|
|
|
|---|
Studies have demonstrated that primary cultures of adipose stromal cells (ASCs) isolated from patients undergoing elective abdominal surgery, express 11ß-HSD1 but not 11ß-HSD2. Activity studies show conversion of inactive cortisone to active cortisol through the expression of 11ß-HSD1 which is significantly higher in omental fat than in subcutaneous fat (Bujalska et al. 1997). Cortisol and insulin treatment were shown to increase differentiation of preadipocytes to adipocytes (Bujalska et al. 1999). Exposure to cortisol also increased expression and activity of 11ß-HSD1, providing a fast-forward feedback system for the local generation of active glucocorticoid within omental adipose tissue (Bujalska et al. 1997). In vivo, such a mechanism would ensure a constant exposure of active glucocorticoid specifically to omental adipose tissue, suggesting that central obesity may reflect ‘Cushing’s disease of the omentum’ (Bujalska et al. 1997). However, in vivo studies are less clear-cut with increased (Paulmyer-Lacroix et al. 2002, Lindsay et al. 2003), unchanged or even reduced 11ß-HSD1 activity reported in obesity. Our own data are consistent with a reduction in hepatic 11ß-HSD1 reductase activity with increasing body mass index (BMI) (Stewart et al. 1999, Rask et al. 2001). Expression in adipose tissue in obesity is more contentious with increased 11ß-HSD1 expression and/or activity again reported in subcutaneous sites. Our data in omental fat suggests no change or even a slight reduction similar to that seen in the liver (Tomlinson et al. 2002). Conversely, enzyme expression increased in obese subjects on a strict calorie-restricted diet (Tomlinson et al. 2004).
Obesity is thought to result from an expansion of adipose tissue mass by hypertrophy of existing adipocytes by lipid accumulation, differentiation of preadipocytes (adipose stromal cells (ASC)) through to adipocytes, or proliferation of preadipocytes (Yau et al. 1995, Tomlinson & Stewart 2001). Cortisol has been shown to cause differentiation of preadipocytes to adipocytes (Hauner et al. 1987), and the expression of 11ß-HSD1 in vitro is sufficient to allow differentiation of omental preadipocytes by cortisone alone (Bujalska et al. 1999). This is supported by inhibition of 11ß-HSD1 with glycerrhetinic acid which prevented cortisone-induced adipocyte differentiation. However, glucocorticoids generally inhibit cellular proliferation, and 11ß-HSDs can regulate this process (Rabbitt et al. 2002). In addition, our group has recently demonstrated inhibition of human omental preadipocyte proliferation with cortisol, modulated by 11ß-HSD1. Thus, in whole adipose tissue, 11ß-HSD1 activity would lead to increased adipocyte differentiation, but simultaneous inhibition of preadipocyte proliferation, and thus the overall impact upon adipose tissue mass needs to be evaluated (Tomlinson & Stewart 2001). Our latest data, supported by data shown in the 11ß-HSD1 KO mouse (Morton et al. 2004), suggest that reduced expression of 11ß-HSD1 in liver and fat might represent an important protective mechanism offsetting the deleterious metabolic consequences of increasing BMI (i.e. reduced hepatic glucose output, reduced adipocyte differentiation). Of interest is that a BMI-related reduction in 11ß-HSD1 activity is not observed in type 2 diabetes mellitus (Valsamakis et al. 2004). Further prospective studies are required to evaluate the consequences of this upon the development of diabetes in obese subjects.
An adipose-specific 11ß-HSD1 transgene has been used to further in vivo studies of the role of 11ß-HSD1 in visceral obesity. 11ß-HSD1 cDNA was linked to the adipocyte fatty-acid-binding protein (aP2) promoter (Masuzaki et al. 2001), which caused a sevenfold amplification of 11ß-HSD1, leading to a viscerally obese phenotype (transgene mice 16% heavier than wild-type mice after 15 weeks). Significantly, this caused a 15–30% elevation in local adipose tissue corticosterone levels but circulating levels remained normal. Adipocyte number remained the same between transgenic and wild-type mice, although exposure of adipocytes to increased glucocorticoid levels resulted in lipid accumulation and an increased adipocyte size in transgenic mice. The transgenic mice were also markedly insulin resistant and glucose intolerant (Masuzaki et al. 2001).
Understandably, these in vitro, clinical and recombinant mouse studies have been the impetus for several pharmaceutical companies to develop selective 11ß-HSD1 inhibitors (selective in that they do not inhibit the related 11ß-HSD2). Many pharmaceutical companies have already published encouraging results using such inhibitors, which are discussed below, and many more are currently investigating newly developed compounds.
Biovitrum-Amgen has shown that arylsulphonamidothiazole compounds inhibit 11ß-HSD1 both in vivo and in vitro (Alberts et al. 2002, Barf et al. 2002) and have shown encouraging results in animal studies. The diethylamide derivative was shown to inhibit human 11ß-HSD1 with an IC50 of 52 nM, and an N-methylpiperazinamide form (BVT.2733) was shown to be specific for the mouse enzyme (IC50 of 96 nM). Both compounds showed >200-fold selectivity over human and murine 11ß-HSD2 (Barf et al. 2002). In the hyperglycaemic mouse strain KKA(y), this compound lowered hepatic PEPCK and glucose-6-phosphatase mRNA, blood glucose and serum insulin concentrations, supporting data that 11ß-HSD1 is a key regulator of gluconeogenesis. This raises the possibility that inhibition of 11ß-HSD1 might be used therapeutically to treat patients with insulin resistance.
Merck has shown that insulin sensitising thiazolidinediones peroxisome proliferator-activated receptor (PPAR)- agonists may mediate their action, in part, through inhibition of adipose 11ß-HSD1, an effect demonstrated in cultured adipocytes (Berger et al. 2001). It was shown that thiazolidinedione and nonthiazolidinedione agonists of PPAR markedly inhibit expression of 11ß-HSD1 in 3T3-L1 adipocytes. This decrease in expression correlated with a significant decrease in the cellular conversion of cortisone to active cortisol. The half-maximal inhibitory effect of the thiazolidinedione, rosiglitazone, occurred at a concentration that supported a PPAR-mediated mechanism of action. It was also demonstrated that the inhibitory action of PPAR agonists on 11ß-HSD1 mRNA expression appears to take place at the level of transcription. In addition, treatment of diabetic db/db mice with rosiglitazone inhibited expression of 11ß-HSD1 in adipose tissue. This decrease in enzyme expression correlated with a significant decline in plasma corticosterone levels (Berger et al. 2001). Furthermore, the lipid lowering agent fenofibrate (a PPAR- agonist) inhibits 11ß-HSD1 in hepatocytes (Hermanowski-Vosatka et al. 2000).
More recently, Merck has identified a new 11ß-HSD1 specific inhibitor – adamantly triazole, and have shown that chronic oral administration in a murine model system lowered triglycerides, insulin, fasting glucose and reduced body weight (Thieringer et al. 2004). This suggests pharmacological inhibition of 11ß-HSD1 improves several key features of the metabolic syndrome.
Glaucoma
11ß-HSD isozyme expression has recently been described in the human and rodent eye (Stokes et al. 2000, Rauz et al. 2001, Suzuki et al. 2001), notably 11ß-HSD1 in ciliary epithelial cells, raising the possibility for a role in aqueous humour production and the regulation of intraocular pressure. Within the aqueous humour, cortisol concentrations exceed cortisone by approximately 14-fold (Rauz et al. 2001). This is highly suggestive of predominantly 11ß-HSD1 reductase activity. Treatment with carbenoxolone in a randomised, double-blind placebo-controlled study of glaucoma sufferers significantly reduced intra-ocular pressure (Rauz et al. 2001) and topical 11ß-HSD1 inhibitors may represent a future novel therapeutic strategy in glaucoma.
Glucocorticoid-induced osteoporosis
Glucocorticoid-induced osteoporosis (GIOP) was first described as a recognised feature of Cushing’s syndrome (Cushing 1932). Since then the increasing use of pharmacological doses of corticosteroids has brought attention to GIOP (Canalis 1996). Studies in vivo clearly show that administration of pharmacological doses of glucocorticoids stimulate bone resorption and inhibit bone formation, therefore decreasing bone mass and increasing the risk of fractures, and causing profound effects on bone cell replication, differentiation and function (Canalis & Delany 2002).
Several studies have demonstrated GR expression in osteoclasts and osteoblastic cells (Chen et al. 1977); GR and MR are present in osteoclasts and osteoblasts (Beavan et al. 2001) and it is thought that many of the effects of glucocorticoids on bone are GR mediated. Studies by our group have demonstrated the expression of 11ß-HSD isozymes in bone. Predominant 11ß-HSD1 expression was demonstrated by enzyme activity studies, RT-PCR and immunohistochemistry, in osteoblast primary cultures (Bland et al. 1999), cell lines and normal adult bone (osteoblasts and osteoclasts) (Cooper et al. 2000). It is proposed that the expression of 11ß-HSD1 in osteoblasts facilitates the local synthesis of active glucocorticoids with consequent effects on osteoblastic proliferation and differentiation. In contrast to liver and adipose tissue, where reductase activity predominates, both reductase and dehydrogenase activities of 11ß-HSD1 are evident in bone chips and primary cultures of human osteoblasts (Cooper et al. 2000).
and MR are present in osteoclasts and osteoblasts (Beavan et al. 2001) and it is thought that many of the effects of glucocorticoids on bone are GR mediated. Studies by our group have demonstrated the expression of 11ß-HSD isozymes in bone. Predominant 11ß-HSD1 expression was demonstrated by enzyme activity studies, RT-PCR and immunohistochemistry, in osteoblast primary cultures (Bland et al. 1999), cell lines and normal adult bone (osteoblasts and osteoclasts) (Cooper et al. 2000). It is proposed that the expression of 11ß-HSD1 in osteoblasts facilitates the local synthesis of active glucocorticoids with consequent effects on osteoblastic proliferation and differentiation. In contrast to liver and adipose tissue, where reductase activity predominates, both reductase and dehydrogenase activities of 11ß-HSD1 are evident in bone chips and primary cultures of human osteoblasts (Cooper et al. 2000).
Tissue damage by pro-inflammatory cytokines is attenuated at both systemic and cellular levels by anti-inflammatory factors such as glucocorticoids. Within the human osteosarcoma cell-line (MG-63) pro-inflammatory cytokines, interleukin (IL)-1ß and tumour necrosis factor (TNF)- potently inhibited 11ß-HSD2 activity, and reciprocally raised 11ß-HSD1 activity and expression. In addition, a rise in 11ß-HSD1 reductase activity was observed in primary cultures of osteoblasts treated with TNF- (Cooper et al. 2001). These data suggest that local tissue metabolism of glucocorticoids is likely to be important in determining the sensitivity of both osteoblasts and osteoclasts to glucocorticoids in normal bone homeostasis. In addition, variation in 11ß-HSD isozyme expression and activity may explain individual differences in susceptibility to glucocorticoid-induced osteoporosis.
Importantly, 11ß-HSD1 reductase activity in osteo-blast cultures has been shown to correlate positively with age. Glucocorticoid treatment caused a time- and dose-dependent increase in 11ß-HSD1 activity with similar increases in 11ß-HSD1 mRNA expression (Cooper et al. 2002). These studies indicate that activation of glucocorticoids at an autocrine level within bone is likely to play an important role in the age-related decrease in bone formation and increased risk of glucocorticoid-induced osteoporosis (Cooper et al. 2002).
| Polycystic ovary syndrome |
|---|
|
|
|---|
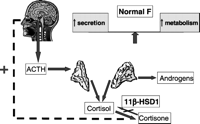
Over 10 years ago, our group postulated that defective 11ß-HSD1 activity may be an important mechanism in patients with PCOS (Stewart et al. 1990). The resulting increase in cortisol metabolic clearance rate would lead to stimulation of the HPA axis in an attempt to normalise serum cortisol levels, and might account for the ACTH-mediated hyperandrogenism in such patients. Alterations of peripheral metabolism of adrenal steroids, specifically increased 5-reductase and 11ß-HSD isozyme activities, have been identified in women with PCOS (Stewart et al. 1990, Rodin et al. 1994, Chin et al. 2000, Moghettii et al. 2002, Fassnacht et al. 2003, Tomlinson et al. 2003). This is perhaps best exemplified through patients with the opposite of AME – ‘cortisone reductase deficiency’.
Cortisone reductase deficiency
The existence of a deficiency in the peripheral enzymatic conversion of E to F (the opposite of AME) was first described in 1984 by Taylor et al., and has since been described in eleven individuals, all but one being female (Taylor et al. 1984, Savage 1991, Nikkila et al. 1993, Phillipov et al. 1996, Jamieson et al. 1999, Nordenstrom et al. 1999). The clinical characteristics of these patients are summarised in Table 3![]() . This rare condition named ‘cortisone reductase deficiency’ (CRD) is a form of PCOS in which the underlying defect appears to be deficient activity of the 11ß-HSD1 enzyme. Patients with CRD share many clinical characteristics associated with PCOS and have invariably presented in adolescence or early adulthood with features of hyperandrogenism (acne, hirsutism, oligo-amenorrhoea, infertility). Obesity has been a feature of some cases. Serum androgens (dehydroandrosterone sulphate (DHEAS), androstene-dione and testosterone) have been elevated in each case, but are readily suppressed following dexamethasone administration.
. This rare condition named ‘cortisone reductase deficiency’ (CRD) is a form of PCOS in which the underlying defect appears to be deficient activity of the 11ß-HSD1 enzyme. Patients with CRD share many clinical characteristics associated with PCOS and have invariably presented in adolescence or early adulthood with features of hyperandrogenism (acne, hirsutism, oligo-amenorrhoea, infertility). Obesity has been a feature of some cases. Serum androgens (dehydroandrosterone sulphate (DHEAS), androstene-dione and testosterone) have been elevated in each case, but are readily suppressed following dexamethasone administration.
|
-HSD1 activity). The defect in E to F conversion results in an increased metabolic clearance rate for cortisol; through the negative feedback mechanism the HPA axis is activated and ACTH secretion is increased to maintain normal circulating cortisol concentrations, but at the expense of ACTH-mediated androgen excess. This leads to a polycystic ovarian syndrome-like phenotype in females (Biason-Lauber et al. 2000), as summarised in Fig. 4
|
Recently, we have reported an extensive genetic investigation of HSD11B1 in three CRD kindreds, each with a single affected case. The CRD cases, one of which is male, all exhibited characteristically low urinary THF+allo-THF:THE ratios of between 0.03 and 0.04 (reference range 0.7–1.1), and have been described previously in the literature (Jamieson et al. 1999, Laing et al. 2002, Malunowicz et al. 2003). In all three CRD cases, two polymorphisms in complete linkage disequilibrium within intron 3 of HSD11B1 were identified; an A insertion (83557), and 40 base pairs downstream a T to G substitution (83597) (Fig. 3A![]() ). The allele frequency for the 83557A/83597T-G haplotype was 14% in control populations.
). The allele frequency for the 83557A/83597T-G haplotype was 14% in control populations.
Although the 11ß-HSD1 cDNA sequence was normal in one case, adipose tissue mRNA levels were 28-fold lower when compared with an unaffected sister, and the corresponding oxo-reductase activity was absent (0% conversion of E to F versus 14% in the unaffected sibling). In luciferase reporter assays, there was a 2.5-fold reduction in transcriptional activity in HSD11B1 constructs containing the intron 3 83557A/83597T-G mutation compared with wild-type, suggesting that this region of the gene acts as an intronic enhancer of HSD11B1 expression. These data are in keeping with published precedents suggesting a silencer/enhancer role for intronic sequences in many genes including HSD11B2 (Agarwal 2001). The impact of heterozygosity for the 83557A/83597T-G mutation upon cortisol metabolism is unknown, but the CRD phenotype cannot be explained by heterozygosity or homozygosity at this single locus because this was present in 25% and 3% of normals respectively.
As discussed previously, 11ß-HSD1 oxo-reductase activity requires NADPH. The purified enzyme behaves as an NADP-dependent dehydrogenase and the switch to oxo-reductase activity upon tissue homogenization (Agarwal et al. 1990) suggests a close association between 11ß-HSD1 and a NADPH generation system. The G6 PDH enzyme of the pentose phosphate pathway has been considered to be the major source of intracellular NADPH (Kletzien et al. 1994), but this is a cytosolic enzyme and the membrane-binding domain of 11ß-HSD1 directs the active site of the enzyme towards the ER lumen, away from the cytosol (Ozols 1995). Here H6 PDH, an enzyme of previously uncertain significance but related to G6 PDH, can catalyze the first two steps of the pentose phosphate pathway and generate NADPH (Kimura et al. 1979, Stegeman & Klotz 1979). H6 PDH is present in most tissues but is highly expressed in liver and adipose tissue, sites of 11ß-HSD1 oxo-reductase activity.
Sequencing of the H6PD gene, localized to chromosome 1p36.2 (see Mason et al. 1999), revealed mutations within exon 5 in all three CRD cases. Case 1 was heterozygous for 620 ins29 bp621, an insert of 29 bp between residues 620 and 621 that results in the inclusion of 3 new amino acids and a stop codon, truncating the protein by 171 amino acids. Cases 2 and 3 were homozygous for R453Q, a non-conservative missense amino acid change. The position of the mutations within H6 PDH are shown in Fig. 3B![]() . When H6PD mutant cDNAs were synthesized and expressed in hepatic WRL68 cells, the 620 ins29 bp621 mutant was devoid of H6 PDH activity and the R453Q mutant demonstrated residual activity that was consistently less than 50% of wild-type. The impact of the R453Q mutation in the context of a normal HSD11B1 genotype is unknown. The allele frequency of R453Q in both Scottish and Indo-Asian controls is 21–22%, so that ~4% of the normal population is homozygous for this mutation. The combination of homozygosity for the H6PD R453Q and heterozygosity for the HSD11B1 intron 3 83557A/ 83597T-G mutations was not observed in our control subjects, and its presence in 2/3 CRD patients is unlikely to be due to chance (P=0.0008, Fisher’s exact test). Based on our normative allelic frequency data we can predict a prevalence rate for CRD of approximately 0.1% for the mutations HSD11B1 +/– and R453Q H6PD –/–. Alternatively, interaction with an unidentified third locus, modifying penetrance, might be implicated.
. When H6PD mutant cDNAs were synthesized and expressed in hepatic WRL68 cells, the 620 ins29 bp621 mutant was devoid of H6 PDH activity and the R453Q mutant demonstrated residual activity that was consistently less than 50% of wild-type. The impact of the R453Q mutation in the context of a normal HSD11B1 genotype is unknown. The allele frequency of R453Q in both Scottish and Indo-Asian controls is 21–22%, so that ~4% of the normal population is homozygous for this mutation. The combination of homozygosity for the H6PD R453Q and heterozygosity for the HSD11B1 intron 3 83557A/ 83597T-G mutations was not observed in our control subjects, and its presence in 2/3 CRD patients is unlikely to be due to chance (P=0.0008, Fisher’s exact test). Based on our normative allelic frequency data we can predict a prevalence rate for CRD of approximately 0.1% for the mutations HSD11B1 +/– and R453Q H6PD –/–. Alternatively, interaction with an unidentified third locus, modifying penetrance, might be implicated.
Thus, a combination of mutations in the HSD11B1 and H6PD genes interact to cause CRD manifesting in a reduction in 11ß-HSD1 expression and impaired provision of NADPH to an enzyme that is critically dependent on reduced co-factor for oxo-reductase activity. This is summarised in diagrammatic form in Fig. 5![]() . A digenic triallelic mode of inheritance is proposed, in which three distinct alleles, from two (or more) loci (HSD11B1 and H6PD), are necessary for trait manifestation (Fig. 3A and B
. A digenic triallelic mode of inheritance is proposed, in which three distinct alleles, from two (or more) loci (HSD11B1 and H6PD), are necessary for trait manifestation (Fig. 3A and B![]() ).
).
|
| Conclusions |
|---|
|
|
|---|
11ß-HSD2, being a unidirectional dehydrogenase, controls renal cortisol concentrations and, via its pre-receptor role, protects the MR from illicit activation by cortisol. Mutations in the gene encoding 11ß-HSD2 lead to the monogenic form of hypertension – AME. It has been suggested that mild mutations in HSD11B2 may account for some cases of idiopathic hypertension.
More recently, attention has focused on the 11ß-HSD1 isoform and its role in human disease states, in particular features of the metabolic syndrome such as obesity and insulin resistance, and also glucocorticoid-related diseases such as osteoporosis and glaucoma. Thus far, studies have predominantly concentrated upon the liver and adipose tissue and the role of 11ß-HSD1 in regulating hepatic gluconeogenesis and fat mass. Obviously, this makes selective inhibitors of 11ß-HSD1 an attractive proposition for the pharmaceutical industry, who are currently rigorously searching for, and developing, 11ß-HSD1 inhibitors with the aim of finding a therapeutic target for the metabolic syndrome. In addition to the clear clinical impact of these inhibitors, they will also be crucial for increasing our understanding of the role of 11ß-HSD1 in vivo. Some caution should be considered here, as these inhibitors are likely to be systemic, and not tissue specific and it will therefore be of importance to tease out the effect of inhibition within individual tissues that show a wide range of 11ß-HSD1 expression. In addition, as many inhibitors are derived from high throughput assays in intact cell systems, it is exciting to speculate that they might be inhibitors of H6 PDH rather than acting directly upon the 11ß-HSD1 enzyme.
The ‘human 11ß-HSD1 knockout’, termed cortisone reductase deficiency has enabled greater understanding of the set-point of activity of 11ß-HSD1. Mutations were found in both HSD11B1 and the gene encoding hexose-6-phosphate dehydrogenase. It is now thought that H6 PDH is responsible for conferring reductase activity upon 11ß-HSD1 by the production of cofactor (NADPH). Further studies are clearly needed to assess the broader impact of regulation of H6 PDH expression and activity as a determinant of 11ß-HSD1 reductase activity and hence autocrine cortisol generation. The next generation of experiments may be to place the last twenty years of 11ß-HSD1 research in the context of H6 PDH activity and expression.
| References |
|---|
|
|
|---|
Agarwal AK, Monder C, Eckstein B & White PC 1989 Cloning and expression of rat cDNA encoding corticosteroid 11ß-dehydrogenase. Journal of Biological Chemistry 264 18939–18943.
Agarwal AK, Tusie-Luna MT, Monder C & White PC 1990 Expression of 11ß-hydroxysteroid dehydrogenase using recombinant Vaccinia virus. Molecular Endocrinology 4 1827–1832.
Agarwal AK, Mune T, Monder C & White PC 1994 NAD+-dependent isoform of 11ß-hydroxysteroid dehydrogenase. Cloning and characterization of cDNA from sheep kidney. Journal of Biological Chemistry 269 25959–25962.
Agarwal AK, Mune T, Monder C & White PC 1995a Mutations in putative glycosylation sites of rat 11ß-hydroxysteroid dehydrogenase affect enzymatic activity. Biochimica et Biophysica Acta 1248 70–74.[CrossRef][Medline]
Agarwal AK, Rogerson FM, Mune T & White PC 1995b Analysis of the human gene encoding the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. Journal of Steroid Biochemistry and Molecular Biology 55 473–479.[CrossRef][Web of Science][Medline]
Agarwal AK, Giacchetti G, Lavery G, Nikkila H, Palermo M, Ricketts M, McTernan C, Bianchi G, Manunta P, Strazzullo P, Mantero F, White PC & Stewart PM 2000 CA-repeat polymorphism in intron 1 of HSD11B2: effects on gene expression and salt sensitivity. Hypertension 36 187–194.
Alberts P, Engblom L, Edling N, Forsgren M, Klingstrom G, Larsson C, Ronquist-Nii Y, Ohman B & Abrahmsen L 2002 Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia 45 1528–1532.[CrossRef][Web of Science][Medline]
Albiston AL, Obeyesekere VR, Smith RE & Krozowski ZS 1994 Cloning and tissue distribution of the human 11ß-hydroxysteroid dehydrogenase type 2 enzyme. Molecular and Cellular Endocrinology 105 R11–R17.[CrossRef][Web of Science][Medline]
Amelung D, Hubener HJ, Rocka L & Meyerheim G 1953 Conversion of cortisone to compound F. Journal of Clinical Endocrinology and Metabolism 13 1125–1126.
Arriza JL, Weinberger C, Corelli G, Glaser TM, Handelin BL, Houseman DE & Evans RM 1987 Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with glucocorticoid receptor. Science 237 268–275.
Arriza JL, Simerly RB, Swanson LW & Evans RM 1988 The neuronal mineralocorticoid receptor as a mediator of glucocorticoid response. Neuron 1 887–900.[CrossRef][Web of Science][Medline]
Axelrod LR 1953 The separation and identification of mixtures of C21 and C19 steroids by paper chromatography. Journal of Biological Chemistry 205 173–184.
Baggia S, Albrecht ED, Babischkin JS & Pepe GJ 1990 Interconversion of cortisol and cortisone in baboon trophoblast and decidua cells in culture. Endocrinology 127 1735–1741.
Barf T, Vallgarda J, Emond R, Haggstrom C, Kurz G, Nygren A, Larwood V, Mosialou E, Axelsson K, Olsson R, Engblom L, Edling N, Ronquist-Nii Y, Ohman B, Alberts P & Abrahmsen L 2002 Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11 beta-hydroxysteroid dehydrogenase type 1. Journal of Medicinal Chemistry 45 3813–3815.[CrossRef][Web of Science][Medline]
Beavan S, Horner A, Bord S, Ireland D & Compston J 2001 Colocalization of glucocorticoid and mineralocorticoid receptors in human bone. Journal of Bone and Mineral Research 16 1496–1504.[CrossRef][Web of Science][Medline]
Berger J, Tanen M, Elbrecht A, Hermanowski-Vosatka A, Moller DE, Wright SD & Thieringer R 2001 Peroxisome proliferator-activated receptor-gamma ligands inhibit adipocyte 11 beta-hydroxysteroid dehydrogenase type 1 expression and activity. Journal of Biological Chemistry 276 12629–12635.
Bernal AL, Flint AP, Anderson AB & Turnbull AC 1980 11 Beta-hydroxysteroid dehydrogenase activity (E.C. 1.1.1.146) in human placenta and decidua. Journal of Steroid Biochemistry 13 1081–1087.[CrossRef][Web of Science][Medline]
Biason-Lauber A, Suter SL, Shackleton CH & Zachmann M 2000 Apparent cortisone reductase deficiency: a rare cause of hyperandrogenemia and hypercortisolism. Hormone Research 53 260–266.[CrossRef][Web of Science][Medline]
Bland R, Worker CA, Noble BS, Eyre LJ, Bujalska IJ, Sheppard MC, Stewart PM & Hewison M 1999 Characterization of 11 beta-hydroxysteroid dehydrogenase activity and corticosteroid receptor expression in human osteosarcoma cell lines. Journal of Endocrinology 161 455–464.[Abstract]
Blum A, Martin HJ & Maser E 2000a Human 11 beta-hydroxysteroid dehydrogenase 1/carbonyl reductase: recombinant expression in the yeast Pichia pastoris and Escherichia coli. Toxicology 144 113–120.
Blum A, Martin HJ & Maser E 2000b Human 11 beta-hydroxysteroid dehydrogenase type 1 is enzymatically active in its nonglycosylated form. Biochemical and Biophysical Research Communications 276 428–434.[CrossRef][Web of Science][Medline]
Brand E, Kato N, Chatelain N, Krozowski ZS, Jeunemaitre X, Corvol P, Plouin PF, Cambien F, Pascoe L & Soubrier F 1998 Structural analysis and evaluation of the 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta-HSD2) gene in human essential hypertension. Journal of Hypertension 16 1627–1633.[CrossRef][Web of Science][Medline]
Brem AS, Bina RB, King T & Morris DJ 1995 Bidirectional activity of 11 beta-hydroxysteroid dehydrogenase in vascular smooth muscle cells. Steroids 60 406–410.
Brown RW, Chapman KE, Edwards CRW & Seckl JR 1993 Human placental 11ß-hydroxysteroid dehydrogenase: evidence for and partial purification of a distinct NAD-dependent isoform. Endocrinology 132 2614–2621.
Brown RW, Chapman KE, Kotelevtsev Y, Yau JL, Lindsay RS, Brett L, Leckie C, Murad P, Lyons V, Mullins JJ, Edwards CR & Seckl JR 1996 Cloning and production of antisera to human placental 11 beta-hydroxysteroid dehydrogenase type 2. Biochemical Journal 313 1007–1017.[Web of Science][Medline]
Bujalska IJ, Kumar S & Stewart PM 1997 Does central obesity reflect ‘Cushing’s disease of the omentum’? Lancet 349 1210–1213.[CrossRef][Web of Science][Medline]
Bujalska IJ, Kumar S, Hewison M & Stewart PM 1999 Differentiation of adipose stromal cells: the roles of glucocorticoids and 11 beta-hydroxysteroid dehydrogenase. Endocrinology 140 3188–3196.
Burton PJ & Waddell BJ 1994 11ß-Hydroxysteroid dehydrogenase in the rat placenta: developmental changes and the effects of altered glucocorticoid exposure. Journal of Endocrinology 143 505–513.
Burton RB, Keutmann EH, Waterhouse C & Schluer EA 1953 The conversion of cortisone acetate to other alphaketolic steroids. Journal of Clinical Endocrinology and Metabolism 13 48–63.
Bush IE 1969 11ß-Hydroxysteroid dehydrogenase: contrast between studies in vivo and studies in vitro. Advances in Biosciences 3 23–39.
Bush IE, Hunter SA & Meigs RA 1968 Metabolism of 11-oxygenated steroids. Metabolism in vitro by preparations of liver. Biochemical Journal 107 239–258.[Web of Science][Medline]
Canalis E 1996 Clinical review 83. Mechanisms of glucocorticoid action in bone: implications to glucocorticoid-induced osteoporosis. Journal of Clinical Endocrinology and Metabolism 81 3441–3447.[CrossRef][Web of Science][Medline]
Canalis E & Delany AM 2002 Mechanisms of glucocorticoid action in bone. Annals of the New York Academy of Sciences 966 73–81.[Web of Science][Medline]
Chen TL, Aronow L & Feldman D 1977 Glucocorticoid receptors and inhibition of bone cell growth in primary culture. Endocrinology 100 619–628.
Chin D, Shackleton C, Prasad VK, Kohn B, David R, Imperato-McGinley J, Cohen H, McMahon DJ & Oberfield SE 2000 Increased 5 alpha-reductase and normal 11 beta-hydroxysteroid dehydrogenase metabolism of C19 and C21 steroids in a young population with polycystic ovarian syndrome. Journal of Pediatric Endocrinology and Metabolism 13 253–259.
Condon J, Ricketts ML, Whorwood CB & Stewart PM 1997 Ontogeny and sexual dimorphic expression of mouse type 2 11ß-hydroxysteroid dehydrogenase. Molecular and Cellular Endocrinology 127 121–128.[CrossRef][Web of Science][Medline]
Cooper MS, Walker EA, Bland R, Fraser WD, Hewison M & Stewart PM 2000 Expression and functional consequences of 11 beta-hydroxysteroid dehydrogenase activity in human bone. Bone 27 375–381.[CrossRef][Web of Science][Medline]
Cooper MS, Bujalska I, Rabbitt E, Walker EA, Bland R, Sheppard MC, Hewison M & Stewart PM 2001 Modulation of 11 beta-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. Journal of Bone Mineral Research 16 1037–1044.
Cooper MS, Rabbitt EH, Goddard PE, Bartlett WA, Hewison M & Stewart PM 2002 Osteoblastic 11 beta-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. Journal of Bone Mineral Research 17 979–986.
Cope CL & Black E 1958 The production rate of cortisol in man. Lancet 14 1020–1024.
Coulter CL, Smith RE, Stowasser M, Sasano H, Krozowski ZS & Gordon RD 1999 Expression of 11 beta-hydroxysteroid dehydrogenase type 2 (11 betaHSD-2) in the developing human adrenal gland and human adrenal cortical carcinoma and adenoma. Molecular and Cellular Endocrinology 154 71–77.[CrossRef][Web of Science][Medline]
Cushing H 1932 The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). Bulletin of the Johns Hopkins Hospital 50 137–195.
Dave-Sharma S, Wilson RC, Harbison MD, Newfield R, Azar MR, Krozowski ZS, Funder JW, Shackleton CH, Bradlow HL, Wei JQ, Hertecant J, Moran A, Neiberger RE, Balfe JW, Fattah A, Daneman D, Akkurt HI, De Santis C & New MI 1998 Examination of genotype and phenotype relationships in 14 patients with apparent mineralocorticoid excess. Journal of Clinical Endocrinology and Metabolism 83 2244–2254.
Despres JP, Moorjani S, Ferland M, Tremblay A, Lupien PJ, Nadeau A, Pinault S, Theriault G & Bouchard C 1989 Adipose tissue distribution and plasma lipoprotein levels in obese women. Importance of intra-abdominal fat. Arteriosclerosis 9 203–210.
Edwards CRW, Stewart PM, Burt D, Brett L, McIntyre MA, Sutanto WS, DeKloet ER & Monder C 1988 Localisation of 11ß-hydroxysteroid dehydrogenase-tissue specific protector of the mineralocorticoid receptor. Lancet ii (8618) 986–989.
Eyre LJ, Rabbitt EH, Bland R, Hughes SV, Cooper MS, Sheppard MC, Stewart PM & Hewison M 2001 Expression of 11 beta-hydroxysteroid dehydrogenase in rat osteoblastic cells: pre-receptor regulation of glucocorticoid responses in bone. Journal of Cellular Biochemistry 81 453–462.[CrossRef][Web of Science][Medline]
Fassnacht M, Schlenz N, Schneider SB, Wudy SA, Allolio B & Arlt W 2003 Beyond adrenal and ovarian androgen generation - increased peripheral 5 alpha-reductase activity in women with polycystic ovary syndrome. Journal of Clinical Endocrinology and Metabolism 88 2760–2766.
Ferrari P, Obeyesekere VR, Li K, Wilson RC, New MI, Funder JW & Krozowski ZS 1996 Point mutations abolish 11ß-hydroxysteroid dehydrogenase type II activity in three families with the congenital syndrome of apparent mineralocorticoid excess. Molecular and Cellular Endocrinology 119 21–24.[CrossRef][Web of Science][Medline]
Ferrari P, Sansonnens A, Dick B & Frey FJ 2001 In vivo 11 beta-HSD-2 activity: variability, salt-sensitivity, and effect of licorice. Hypertension 38 1330–1336.
Fujioka S, Matsuzawa Y, Tokunaga K & Tarui S 1987 Contribution of intra-abdominal fat accumulation to the impairment of glucose and lipid metabolism in human obesity. Metabolism 36 54–59.[CrossRef][Web of Science][Medline]
Hauner H, Schmid P & Pfeiffer EF 1987 Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. Journal of Clinical Endocrinology and Metabolism 64 832–835.
Hellman L, Nakada F, Zumoff B, Fukushima D, Bradlow HL & Gallagher TF 1971 Renal capture and oxidation of cortisol in man. Journal of Clinical Endocrinology and Metabolism 33 52–62.
Hermanowski-Vosatka A, Gerhold D, Mundt SS, Loving VA, Lu M, Chen Y, Elbrecht A, Wu M, Doebber T, Kelly L, Milot D, Guo Q, Wang PR, Ippolito M, Chao YS, Wright SD & Thieringer R 2000 PPARalpha agonists reduce 11 beta-hydroxysteroid dehydrogenase type 1 in the liver. Biochemical and Biophysical Research Communications 279 330–336.[CrossRef][Web of Science][Medline]
Hirasawa G, Sasano H, Suzuki T, Takeyama J, Muramatu Y, Fukushima K, Hiwatashi N, Toyota T, Nagura H & Krozowski ZS 1999 11 Beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor in human fetal development. Journal of Clinical Endocrinology and Metabolism 84 1453–1458.
Holmes MC, Kotelevtsev Y, Mullins JJ & Seckl JR 2001 Phenotypic analysis of mice bearing targeted deletions of 11 beta-hydroxysteroid dehydrogenases 1 and 2 genes. Molecular and Cellular Endocrinology 171 15–20.[CrossRef][Web of Science][Medline]
Hundertmark S, Buhler H, Ragosch V, Dinkelborg L, Arabin B & Weitzel HK 1995 Correlation of surfactant phosphatidylcholine synthesis and 11ß-hydroxysteroid dehydrogenase in fetal lung. Endocrinology 136 2573–2578.[Abstract]
Jamieson P, Chapman KE, Edwards CR & Seckl JR 1995 11ß-Hydroxysteroid dehydrogenase is an exclusive 11ß-reductase in primary cultures of rat hepatocytes: effect of physiochemical and hormonal manipulations. Endocrinology 136 4754–4761.[Abstract]
Jamieson A, Wallace AM, Andrew R, Nunez BS, Walker BR, Fraser R, White PC & Connell JM 1999 Apparent cortisone reductase deficiency: a functional defect in 11 beta-hydroxysteroid dehydrogenase type 1. Journal of Clinical Endocrinology and Metabolism 84 3570–3574.
Jenkins JS 1966 The metabolism of cortisol by human extra-hepatic tissues. Journal of Endocrinology 34 51–56.
Kendall EC 1971 Arthritis. In Cortisone pp. 121–137. Charles Scriber’s Sons: New York.
Kendall JW & Allen C 1968 Studies on the glucocorticoid feedback control of ACTH secretion. Endocrinology 82 397–405.
Kenouch S, Lombes M, Delahaye F, Eugene E, Bonvalet JP & Farman N 1994 Human skin as target for aldosterone: coexpression of mineralocorticoid receptors and 11 beta-hydroxysteroid dehydrogenase. Journal of Clinical Endocrinology and Metabolism 79 1334–1341.[Abstract]
Kimura K, Endou H, Sudo J & Sakai F 1979 Glucose dehydrogenase (hexose 6-phosphate dehydrogenase) and the microsomal electron transport system. Evidence supporting their possible functional relationship. Journal of Biochemistry (Tokyo) 85 319–326.
Kitanaka S, Katsumata N, Tanae A, Hibi I, Takeyama K, Fuse H, Kato S & Tanaka T 1997 A new compound heterozygous mutation in the 11ß-hydroxysteroid dehydrogenase type 2 gene in a case of apparent mineralocorticoid excess. Journal of Clinical Endocrinology and Metabolism 82 4054–4058.
Klemcke HG, Sampath KR, Yang K, Vallet JL & Christenson RK 2003 11 Beta-hydroxysteroid dehydrogenase and glucocorticoid receptor messenger RNA expression in porcine placentae: effects of stage of gestation, breed, and uterine environment. Biology of Reproduction 69 1945–1950.
Kletzien RF, Harris PK & Foellmi LA 1994 Glucose-6-phosphate dehydrogenase: a ‘housekeeping’ enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB Journal 8 174–181.[Abstract]
Korbonits M, Bujalska I, Shimojo M, Nobes J, Jordan S, Grossman AB & Stewart PM 2001 Expression of 11 beta-hydroxysteroid dehydrogenase isoenzymes in the human pituitary: induction of the type 2 enzyme in corticotropinomas and other pituitary tumors. Journal of Clinical Endocrinology and Metabolism 86 2728–2733.
Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CR, Seckl JR & Mullins JJ 1997 11 Beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. PNAS 94 14924–14929.
Kotelevtsev Y, Brown RW, Fleming S, Kenyon C, Edwards CR, Seckl JR & Mullins JJ 1999 Hypertension in mice lacking 11 beta-hydroxysteroid dehydrogenase type 2. Journal of Clinical Investigation 103 683–689.[Web of Science][Medline]
Krozowski Z & Funder JW 1983 Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intinsic steroid specificity. PNAS 80 6056–6060.
Krozowski ZS, Maguire JA, Stein-Oakley AN, Dowling J, Smith RE & Andrews RK 1995 Immunohistochemical localization of the 11ß-hydroxysteroid dehydrogenase type II enzyme in human kidney and placenta. Journal of Clinical Endocrinology and Metabolism 80 2203–2209.[Abstract]
Laing I, Adams JE, Wood PJ, Taylor NF & Ray DW 2002 Cortisone reductase deficiency (11 beta-hydroxysteroid dehydrogenase type 1) deficiency presenting with features of late onset congenital adrenal hyperplasia. 21st Joint Meeting of the British Endocrine Societies, 8–11 April 2002.
Lakshmi V & Monder C 1988 Purification and characterization of the corticosteroid 11ß- dehydrogenase component of the rat liver 11ß-hydroxysteroid dehydrogenase complex. Endocrinology l23 2390–2398.
Lakshmi V, Nath N & Muneyyirci-Delale O 1993 Characterisation of 11ß-hydroxysteroid dehydrogenase of human placenta: evidence for the existence of two species of 11ß- hydroxysteroid dehydrogenase. Journal of Steroid Biochemistry and Molecular Biology 45 391–397.[CrossRef][Web of Science][Medline]
Lavery GG, McTernan CL, Bain SC, Chowdhury TA, Hewison M & Stewart PM 2002 Association studies between the HSD11B2 gene (encoding human 11 beta-hydroxysteroid dehydrogenase type 2), type 1 diabetes mellitus and diabetic nephropathy. European Journal of Endocrinology 146 553–558.[Abstract]
Lavery GG, Ronconi V, Draper N, Rabbitt EA, Lyons V, Chapman KE, Walker EA, McTernan CL, Giacchetti G, Mantero F, Seckl JR, Edwards CRW, Conell JMC, Hewison M & Stewart PM 2003 Late onset ‘apparent mineralocorticoid excess’ due to novel compound heterozygous mutations in the HSD11B2 gene. Hypertension 42 123–129.
Leckie CM, Welberg LA & Seckl JR 1998 11 Beta-hydroxysteroid dehydrogenase is a predominant reductase in intact rat Leydig cells. Journal of Endocrinology 159 233–238.[Abstract]
Li A, Li KXZ, Marui S, Krozowski ZS, Batista MC, Whorwood CB, Arnhold IJP, Shackleton CHL, Mendonca BB & Stewart PM 1997 Apparent mineralocorticoid excess in a Brazilian kindred: hypertension in the heterozygote state. Journal of Hypertension 15 1397–1402.[CrossRef][Web of Science][Medline]
Li A, Tedde R, Krozowski ZS, Pala A, Li KX, Shackleton CH, Mantero F, Palermo M & Stewart PM 1998 Molecular basis for hypertension in the ‘type II variant’ of apparent mineralocorticoid excess. American Journal of Human Genetics 63 370–379.[CrossRef][Web of Science][Medline]
Lindsay RS, Wake DJ, Nair S, Bunt J, Livingstone DE, Permana PA, Tataranni PA & Walker BR 2003 Subcutaneous adipose 11 beta-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in Pima Indians and Caucasians. Journal of Clinical Endocrinology and Metabolism 88 2738–2744.
Lin-Su K, Zhou P, Arora N, Betensky BP, New MI & Wilson RC 2004 In vitro expression studies of a novel mutation delta299 in a patient affected with apparent mineralocorticoid excess. Journal of Clinical Endocrinology and Metabolism 89 2024–2027.
Lopez BA & Craft IL 1981 Corticosteroid metabolism in vitro by human placenta, fetal membranes and decidua in early and late gestation. Placenta 2 279–285.
Malunowicz EM, Romer TE, Urban M & Bossowski A 2003 11 Beta-hydroxysteroid dehydrogenase type 1 deficiency (‘apparent cortisone reductase deficiency’) in a 6-year-old boy. Hormone Research 59 205–210.[CrossRef][Web of Science][Medline]
Maser E, Volker B & Friebertshauser J 2002 11 Beta-hydroxysteroid dehydrogenase type 1 from human liver: dimerization and enzyme cooperativity support its postulated role as glucocorticoid reductase. Biochemistry 41 2459–2465.[CrossRef][Web of Science][Medline]
Maser E, Friebertshauser J & Volker B 2003 Purification, characterization and NNK carbonyl reductase activities of 11 beta-hydroxysteroid dehydrogenase type 1 from human liver: enzyme cooperativity and significance in the detoxification of a tobacco-derived carcinogen. Chemico-Biological Interactions 143–144 435–448.
Mason PJ, Stevens D, Diez A, Knight SW, Scopes DA & Vulliamy TJ 1999 Human hexose-6-phosphate dehydrogenase (glucose 1-dehydrogenase) encoded at 1p36: coding sequence and expression. Blood Cells, Molecules, and Diseases 25 30–37.[CrossRef][Web of Science][Medline]
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR & Flier JS 2001 A transgenic model of visceral obesity and the metabolic syndrome. Science 294 2166–2170.
Milford DV, Shackleton CH & Stewart PM 1995 Mineralocorticoid hypertension and congenital deficiency of 11 beta-hydroxysteroid dehydrogenase in a family with the syndrome of ‘apparent’ mineralocorticoid excess. Clinical Endocrinology 43 241–246.[Medline]
Moghettii P, Spiazza GG, Tosi F, Solagna E, Zenari MI, Muggeo M & Castello R 2002 Cortisol secretion and metabolism in PCOS women. 84th Annual Meeting of the Endocrine Society, San Francisco, CA, USA.
Monder C & Lakshmi V 1989 Evidence for kinetically distinct forms of corticosteroid 11ß-dehydrogenase in rat liver microsomes. Journal of Steroid Biochemistry 32 77–83.[CrossRef][Web of Science][Medline]
Monder C & White PC 1993 11 beta-hydroxysteroid dehydrogenase. Vitamins and Hormones 47 187–271.[Web of Science][Medline]
Monder C, Shackleton CHL, Bradlow HL, New MI, Stoner E, Iohan F & Lakshmi V 1986 The syndrome of apparent mineralocorticoid excess: its association with 11ß-dehydrogenase and 5ß-reductase deficiency and some consequences for corticosteroid metabolism. Journal of Clinical Endocrinology and Metabolism 63 550–557.
Monder C, Miroff Y, Marandici A & Hardy M 1994 11ß-Hydroxysteroid dehydrogenase alleviates glucocorticoid-mediated inhibition of steroidogenesis in rat Leydig cells. Endocrinology 134 1199–1204.
Moore CC, Mellon SH, Murai J, Siiteri PK & Miller WL 1993 Structure and function of the hepatic form of 11 beta-hydroxysteroid dehydrogenase in the squirrel monkey, an animal model of glucocorticoid resistance. Endocrinology 133 368–375.
Morineau G, Marc JM, Boudi A, Galons H, Gourmelen M, Corvol P, Pascoe L & Fiet J 1999 Genetic, biochemical, and clinical studies of patients with A328 V or R213C mutations in 11 betaHSD2 causing apparent mineralocorticoid excess. Hypertension 34 435–441.
Morton NM, Ramage L & Seckl JR 2004 Down-regulation of adipose 11 beta-hydroxysteroid dehydrogenase type 1 by high-fat feeding in mice: a potential adaptive mechanism counteracting metabolic disease. Endocrinology 145 2707–2712.
Moudgil A, Rodic G, Jordon SC & Kamil ES 2000 Nephrocalcinosis and renal cysts associated with apparent mineralocorticoid excess syndrome. Pediatric Nephrology 15 60–62.[CrossRef][Web of Science][Medline]
Mune T & White PC 1996 Apparent mineralocorticoid excess; genotype is correlated with biochemical phenotype. Hypertension 27 1193–1199.
Mune T, Rogerson FM, Nikkilä H, Agarwal AK & White PC 1995 Human hypertension caused by mutations in the kidney isozyme of 11ß-hydroxysteroid dehydrogenase. Nature Genetics 10 394–399.[CrossRef][Web of Science][Medline]
Naray-Fejes-Toth A & Fejes-Toth G 1995 Expression cloning of the aldosterone target cell-specific 11 beta-hydroxysteroid dehydrogenase from rabbit collecting duct cells. Endocrinology 136 2579–2586.[Abstract]
Nikkila H, Tannin GM, New MI, Taylor NF, Kalaitzoglou G, Monder C & White PC 1993 Defects in the HSD11 gene encoding 11 beta-hydroxysteroid dehydrogenase are not found in patients with apparent mineralocorticoid excess or 11-oxoreductase deficiency. Journal of Clinical Endocrinology and Metabolism 77 687–691.[Abstract]
Nobel CS, Dunas F & Abrahmsen LB 2002 Purification of full-length recombinant human and rat type 1 11 beta-hydroxysteroid dehydrogenases with retained oxidoreductase activities. Protein Expression and Purification 26 349–356.[CrossRef][Web of Science][Medline]
Nordenstrom A, Marcus C, Axelson M, Wedell A & Ritzen EM 1999 Failure of cortisone acetate treatment in congenital adrenal hyperplasia because of defective 11 beta-hydroxysteroid dehydrogenase reductase activity. Journal of Clinical Endocrinology and Metabolism 84 1210–1213.
Nunez BS, Rogerson FM, Mune T, Igarashi Y, Nakagawa Y, Phillipov G, Moudgil A, Travis LB, Palermo M, Shackleton C & White PC 1999 Mutants of 11 beta-hydroxysteroid dehydrogenase (11-HSD2) with partial activity: improved correlations between genotype and biochemical phenotype in apparent mineralocorticoid excess. Hypertension 34 638–642.
Obeyesekere VR, Trzeciak WH, Li KX & Krozowski ZS 1998 Serines at the active site of 11 beta-hydroxysteroid dehydrogenase type I determine the rate of catalysis. Biochemical and Biophysical Research Communications 250 469–473.[CrossRef][Web of Science][Medline]
Odermatt A, Arnold P, Stauffer A, Frey BM & Frey FJ 1999 The N-terminal anchor sequences of 11 beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. Journal of Biological Chemistry 274 28762–28770.
Odermatt A, Dick B, Arnold P, Zaehner T, Plueschke V, Deregibus MN, Repetto H, Frey BM, Frey FJ & Ferrari P 2001 A mutation in the cofactor-binding domain of 11 beta-hydroxysteroid dehydrogenase type 2 associated with mineralocorticoid hypertension. Journal of Clinical Endocrinology and Metabolism 86 1247–1252.
Osinski PA 1960 Steroid 11ß-ol dehydrogenase in human placenta. Nature 187 777.[CrossRef][Medline]
Ozols J 1995 Lumenal orientation and post-translational modifications of the liver microsomal 11 beta-hydroxysteroid dehydrogenase [published erratum appears in Journal of Biological Chemistry 1995 270 10360]. Journal of Biological Chemistry 270 2305–2312.
Palermo M, Shackleton CHL, Mantero F & Stewart PM 1996 Urinary free cortisone and the assessment of 11ß-hydroxysteroid dehydrogenase activity in man. Clinical Endocrinology 45 605–611.[CrossRef][Medline]
Palermo M, Cossu M & Shackleton CH 1998 Cure of apparent mineralocorticoid excess by kidney transplantation. New England Journal of Medicine 339 1787–1788.
Parks LL, Turney MK, Gaitan D & Kovacs WJ 1998 Expression of 11 beta-hydroxysteroid dehydrogenase type 2 in an ACTH-producing small cell lung cancer. Journal of Steroid Biochemistry and Molecular Biology 67 341–346.[CrossRef][Web of Science][Medline]
Paulmyer-Lacroix O, Boullu S, Oliver C, Alessi MC & Grino M 2002 Expression of the mRNA coding for 11 beta-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: an in situ hybridization study. Journal of Clinical Endocrinology and Metabolism 87 2701–2705.
Pepe GJ, Burch MG & Albrecht ED 1999 Expression of the 11 beta-hydroxysteroid dehydrogenase types 1 and 2 proteins in human and baboon placental syncytiotrophoblast. Placenta 20 575–582.[CrossRef][Medline]
Persson B, Krook M & Jörnvall H 1991 Characteristics of short-chain alcohol dehydrogenases and related enzymes. European Journal of Biochemistry 200 537–543.[Web of Science][Medline]
Phillipou G & Higgins BA 1985 A new defect in the peripheral conversion of cortisone to cortisol. Journal of Steroid Biochemistry 22 435–436.[CrossRef][Web of Science][Medline]
Phillipov G, Palermo M & Shackleton CH 1996 Apparent cortisone reductase deficiency: a unique form of hypercortisolism. Journal of Clinical Endocrinology and Metabolism 81 3855–3860.
Phillips DM, Lakshmi V & Monder C 1989 Corticosteroid 11ß-dehydrogenase in rat testis. Endocrinology 125 209–216.
Pu X & Yang K 2000 Guinea pig 11 beta-hydroxysteroid dehydrogenase type 1: primary structure and catalytic properties. Steroids 65 148–156.
Quinkler M, Bappal B, Draper N, Atterbury AJ, Lavery GG, Walker EA, DeSilva V, Taylor NF, Hala S, Rajendra N & Stewart PM 2004 Molecular basis for the apparent mineralocorticoid excess syndrome in the Oman population. Molecular and Cellular Endocrinology 217 143–149.[CrossRef][Web of Science][Medline]
Rabbitt EH, Lavery GG, Walker EA, Cooper MS, Stewart PM & Hewison M 2002 Prereceptor regulation of glucocorticoid action by 11 beta-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB Journal 16 36–44.
Rajan V, Chapman KE, Lyons V, Jamieson P, Mullins JJ, Edwards CRW & Seckl JR 1995 Cloning, sequencing and tissue distribution of mouse 11ß-hydroxysteroid dehydrogenase-1 cDNA. Journal of Steroid Biochemistry and Molecular Biology 52 141–147.[CrossRef][Web of Science][Medline]
Rajan V, Edwards CR & Seckl JR 1996 11 Beta-hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. Journal of Neuroscience 16 65–70.
Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DE, Johnson O & Walker BR 2001 Tissue-specific dysregulation of cortisol metabolism in human obesity. Journal of Clinical Endocrinology and Metabolism 86 1418–1421.
Rauz S, Walker EA, Shackleton CH, Hewison M, Murray PI & Stewart PM 2001 Expression and putative role of 11 beta-hydroxysteroid dehydrogenase isozymes within the human eye. Investigative Ophthalmology and Visual Science 42 2037–2042.
Ricketts ML, Verhaeg J, Bujalska I, Howie AJ, Rainey WE & Stewart PM 1998 Immunohistological localization of type 1 11ß-hydroxysteroid dehydrogenase in human tissues. Journal of Clinical Endocrinology and Metabolism 83 1325–1335.
Riddle MC & McDaniel PA 1994 Renal 11 beta-hydroxysteroid dehydrogenase activity is enhanced by ramipril and captopril. Journal of Clinical Endocrinology and Metabolism 78 830–834.[Abstract]
Rodin A, Thakkar H, Taylor N & Clayton R 1994 Hyperandrogenism in polycystic ovary syndrome: evidence of dysregulation of 11ß-hydroxysteroid dehydrogenase. New England Journal of Medicine 330 460–465.
Rogatsky I, Trowbridge JM & Garabedian MJ 1997 Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Molecular Cell Biology 17 3181–3193.
Rogoff D, Smolenicka Z, Bergada I, Vallejo G, Barontini M, Heinrich JJ & Ferrari P 1998 The codon 213 of the 11 beta-hydroxysteroid dehydrogenase type 2 gene is a hot spot for mutations in apparent mineralocorticoid excess. Journal of Clinical Endocrinology and Metabolism 83 4391–4393.
Roland BL & Funder JW 1996 Localization of 11 beta-hydroxysteroid dehydrogenase type 2 in rat tissues: in situ studies. Endocrinology 137 1123–1128.[Abstract]
Sanchez I, Goya L, Vallerga AK & Firestone GL 1993 Glucocorticoids reversibly arrest rat hepatoma cell growth by inducing an early G1 block in cell cycle progression. Cell Growth and Differentiation 4 215–225.
Savage MW 1991 Increased metabolic clearance of cortisol in corticosteroid 11-reductase deficiency. Journal of Endocrinology 129S 219.
Scarpitta AM & Sinagra D 2000 Polycystic ovary syndrome: an endocrine and metabolic disease. Gynecological Endocrinology 14 392–395.[Web of Science][Medline]
Shackleton CHL & Stewart PM 1990 The hypertension of apparent mineralocorticoid excess (AME) syndrome. In Endocrine Hypertension, pp 155–173. Eds EG Biglieri & JE Melby. New York: Raven Press Ltd.
Shackleton CHL, Rodriguez J, Arteaga E, Lopez JM & Winter JSD 1985 Congenital 11ß-hydroxysteroid dehydrogenase deficiency associated with juvenile hypertension. Corticosteroid metabolite profiles of four patients and their families. Clinical Endocrinology 22 701–712.[Medline]
Shafqat N, Elleby B, Svensson S, Shafqat J, Jornvall H, Abrahmsen L & Oppermann U 2003 Comparative enzymology of 11 beta-hydroxysteroid dehydrogenase type 1 from glucocorticoid resistant (guinea pig) versus sensitive (human) species. Journal of Biological Chemistry 278 2030–2035.
Shams M, Kilby MD, Somerset DA, Howie A, Gupta A, Wood PJ, Afnan M & Stewart PM 1998 11ß-Hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Human Reproduction 13 799–804.
Smolenicka Z, Bach E, Schaer A, Liechti-Gallati S, Frey BM, Frey FJ & Ferrari P 1998 A new polymorphic restriction site in the human 11 beta-hydroxysteroid dehydrogenase type 2 gene. Journal of Clinical Endocrinology and Metabolism 83 1814–1817.
Soro A, Ingram MC, Tonolo G, Glorioso N & Fraser R 1995 Evidence of coexisting changes in 11ß-hydroxysteroid dehydrogenase and 5ß-reductase activity in patients with untreated essential hypertension. Hypertension 25 67–70.
Sprague RG, Power MH & Mason HL 1950 Physiological effects of cortisone and pituitary adrenocorticotropic hormone (ACTH) in man. Journal of the American Medical Association 144 1341–1347.
Srivastava LS, Werk EE Jr, Thrasher K, Sholiton LJ, Kozera R, Nolten W & Knowles HC Jr 1973 Plasma cortisone concentration as measured by radioimmunoassay. Journal of Clinical Endocrinology and Metabolism 36 937–943.
Stegeman JJ & Klotz AV 1979 A possible role for microsomal hexose-6-phosphate dehydrogenase in microsomal electron transport and mixed-function oxygenase activity. Biochemical and Biophysical Research Communications 87 410–415.[CrossRef][Web of Science][Medline]
Stewart PM 1996 11ß-Hydoxysteroid dehydrogenase: implications for clinical medicine. Clinical Endocrinology 44 493–499.[CrossRef][Medline]
Stewart PM, Corrie JET, Shackleton CHL & Edwards CRW 1988 Syndrome of apparent mineralocorticoid excess: a defect in the cortisol–cortisone shuttle. Journal of Clinical Investigation 82 340–349.[Web of Science][Medline]
Stewart PM, Shackleton CHL, Beastall GH & Edwards CRW 1990 5ß-Reductase activity in polycystic ovary syndrome. Lancet 335 431–433.[CrossRef][Web of Science][Medline]
Stewart PM, Murry BA & Mason JI 1994 Human kidney 11ß-hydroxysteroid dehydrogenase is a high affinity nicotinamide adenine dinucleotide-dependent enzyme and differs from the cloned ‘type I’ isoform. Journal of Clinical Endocrinology and Metabolism 79 480–484.[Abstract]
Stewart PM, Whorwood CB & Mason JI 1995 Type 2 11 beta-hydroxysteroid dehydrogenase in foetal and adult life. Journal of Steroid Biochemistry and Molecular Biology 55 465–471.[CrossRef][Web of Science][Medline]
Stewart PM, Krozowski ZS, Gupta A, Milford DV, Howie AJ, Sheppard MC & Whorwood CB 1996 Hypertension in the syndrome of apparent mineralocorticoid excess due to mutation of the 11ß-hydroxysteroid dehydrogenase type 2 gene. Lancet 347 88–91.[CrossRef][Web of Science][Medline]
Stewart PM, Boulton A, Kumar S, Clark PM & Shackleton CH 1999 Cortisol metabolism in human obesity: impaired cortisone" border="0">cortisol conversion in subjects with central adiposity. Journal of Clinical Endocrinology and Metabolism 84 1022–1027.
Stokes J, Noble J, Brett L, Phillips C, Seckl JR, O’Brien C & Andrew R 2000 Distribution of glucocorticoid and mineralocorticoid receptors and 11 beta-hydroxysteroid dehydrogenases in human and rat ocular tissues. Investigative Ophthalmology and Visual Science 41 1629–1638.
Sun K, Yang K & Challis JR 1997 Differential expression of 11ß-hydroxysteroid dehydrogenase types 1 and 2 in human placenta and fetal membranes. Journal of Clinical Endocrinology and Metabolism 82 300–305.
Suzuki S, Suzuki T, Tsubochi H, Koike K, Tateno H, Krozowski ZS & Sasano H 2000 Expression of 11 beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor in primary lung carcinomas. Anticancer Research 20 323–328.[Web of Science][Medline]
Suzuki T, Sasano H, Kaneko C, Ogawa S, Darnel AD & Krozowski ZS 2001 Immunohistochemical distribution of 11 beta-hydroxysteroid dehydrogenase in human eye. Molecular and Cellular Endocrinology 173 121–125.[CrossRef][Web of Science][Medline]
Tannin GM, Agarwal AK, Monder C, New MI & White PC 1991 The human gene for 11ß-hydroxysteroid dehydrogenase. Structure, tissue distribution, and chromosomal localization. Journal of Biological Chemistry 266 16653–16658.
Taylor NF, Bartlett WA & Dawson DJ 1984 Cortisone reductase deficiency: evidence for a new inborn error in metabolism of adrenal steroids. Journal of Endocrinology 102S 89.
Taylor NF, Pollock A & Dornan TL 2003 Corticosteroid 11-reductase deficiency: steroid studies in a further family. Journal of Clinical Investigation 13 238.
Tetsuka M, Yamamoto S, Hayashida N, Hayashi KG, Hayashi M, Acosta TJ & Miyamoto A 2003 Expression of 11 beta-hydroxysteroid dehydrogenases in bovine follicle and corpus luteum. Journal of Endocrinology 177 445–452.[Abstract]
Thieringer R, Balkovec JM, Chen HY, Frazier E, Le Grand CB, Li Z, Metzger JM, Mundt SS, Nunes CN, Strowski M, Olson SH, Robertson N, Strack AM, Schaeffer JM, Zhang BB & Hermanowski-Vosatka A 2004 A novel non-steroidal inhibitor of 11 beta-hydroxysteroid dehydrogenase type 1 improves features of metabolic syndrome in murine disease models. 23rd Joint Meeting of the British Endocrine Societies with the European Federation of Endocrine Societies, Brighton, UK 22–24 March 2004.
Tomlinson JW & Stewart PM 2001 Cortisol metabolism and the role of 11 beta-hydroxysteroid dehydrogenase. Best Practise and Research. Clinical Endocrinology and Metabolism 15 61–78.
Tomlinson JW, Sinha B, Bujalska I, Hewison M & Stewart PM 2002 Expression of 11 beta-hydroxysteroid dehydrogenase type 1 in adipose tissue is not increased in human obesity. Journal of Clinical Endocrinology and Metabolism 87 5630–5635.
Tomlinson JW, Crabtree N, Clark PM, Holder G, Toogood AA, Shackleton CH & Stewart PM 2003 Low-dose growth hormone inhibits 11 beta-hydroxysteroid dehydrogenase type 1 but has no effect upon fat mass in patients with simple obesity. Journal of Clinical Endocrinology and Metabolism 88 2113–2118.
Tomlinson JW, Moore JS, Clark PM, Holder G, Shakespeare L & Stewart PM 2004 Weight loss increases 11 beta-hydroxysteroid dehydrogenase type 1 expression in human adipose tissue. Journal of Clinical Endocrinology and Metabolism 89 2711–2716.
Ugrasbul F, Wiens T, Rubinstein P, New MI & Wilson RC 1999 Prevalence of mild apparent mineralocorticoid excess in Mennonites. Journal of Clinical Endocrinology and Metabolism 84 4735–4738.
Ulick S, Levine LS, Gunczler P, Zanconato G, Ramirex LC, Rauh W, Rosler A, Bradlow JL & New MI 1979 A syndrome of apparent mineralocorticoid excess associated with defects in the peripheral metabolism of cortisol. Journal of Clinical Endocrinology and Metabolism 49 757–764.
Valsamakis G, Anwar A, Tomlinson JW, Shackleton CH, McTernan PG, Chetty R, Wood PJ, Banerjee AK, Holder G, Barnett AH, Stewart PM & Kumar S 2004 11 Beta-hydroxysteroid dehydrogenase type 1 activity in lean and obese males with type 2 diabetes mellitus. Journal of Clinical Endocrinology and Metabolism 89 4755–4761.
Walker BR, Stewart PM, Shackleton CHL, Padfield PL & Edwards CRW 1993 Deficient inactivation of cortisol by 11ß-hydroxysteroid dehydrogenase in essential hypertension. Clinical Endocrinology 39 221–227.[Medline]
Walker EA, Clark AM, Hewison M, Ride JP & Stewart PM 2001 Functional expression, characterization, and purification of the catalytic domain of human 11 beta-hydroxysteroid dehydrogenase type 1. Journal of Biological Chemistry 276 21343–21350.
White PC, Obeid J, Agarwal AK, Tannin GM & Nikkilä H 1994 Genetic analysis of 11ß-hydroxysteroid dehydrogenase. Steroids 59 111–115.
White PC, Mune T & Agarwal AK 1997 11ß-Hydroxysteroid dehydrogenase and the syndrome of apparent mineralocorticoid excess. Endocrine Reviews 18 135–156.
Whitworth JA, Stewart PM, Burt D, Atherden SM & Edwards CR 1989 The kidney is the major site of cortisone production in man. Clinical Endocrinology 31 355–361.[Medline]
Whorwood CB & Stewart PM 1996 Human hypertension caused by mutations in the 11 beta-hydroxysteroid dehydrogenase gene: a molecular analysis of apparent mineralocorticoid excess. Journal of Hypertension (Supplement) 14 S19–S24.
Whorwood CB, Ricketts ML & Stewart PM 1994 Epithelial cell localization of type 2 11 beta-hydroxysteroid dehydrogenase in rat and human colon. Endocrinology 135 2533–2541.[Abstract]
Whorwood CB, Mason JI, Ricketts ML, Howie AJ & Stewart PM 1995 Detection of human 11 beta-hydroxysteroid dehydrogenase isoforms using reverse-transcriptase-polymerase chain reaction and localization of the type 2 isoform to renal collecting ducts. Molecular and Cellular Endocrinology 110 R7–R12.[CrossRef][Web of Science][Medline]
Wilson RC, Harbison MD, Krozowski ZS, Funder JW, Shackleton CHL, Hanauske-Abel HM, Wei JQ, Hertecant J, Moran A, Neiberger RE, Balfe JW, Fattah A, Daneman D, Licholai T & New MI 1995a Several homozygous mutations in the gene for 11ß-hydroxysteroid dehydrogenase type 2 in patients with apparent mineralocorticoid excess. Journal of Clinical Endocrinology and Metabolism 80 3145–3150.[Abstract]
Wilson RC, Krozowski ZS, Li K, Obeyesekere VR, Razzaghy-Azar M, Harbison MD, Wei JQ, Shackleton CHL, Funder JW & New MI 1995b A mutation in the HSD11B2 gene in a family with apparent mineralocorticoid excess. Journal of Clinical Endocrinology and Metabolism 80 2263–2266.[Abstract]
Wilson RC, Dave-Sharma S, Wei JQ, Obeyesekere VR, Li K, Ferrari P, Krozowski ZS, Shackleton CH, Bradlow L, Wiens T & New MI 1998 A genetic defect resulting in mild low-renin hypertension. PNAS 95 10200–10205.
Yang K, Smith CL, Dales D, Hammond GL & Challis JR 1992 Cloning of an ovine 11 beta-hydroxysteroid dehydrogenase complementary deoxyribonucleic acid: tissue and temporal distribution of its messenger ribonucleic acid during fetal and neonatal development. Endocrinology 131 2120–2126.
Yau JL, Olsson T, Morris RG, Meaney MJ & Seckl JR 1995 Glucocorticoids, hippocampal corticosteroid receptor gene expression and antidepressant treatment: relationship with spatial learning in young and aged rats. Neuroscience 66 571–581.[CrossRef][Medline]
Zhou MY, Gomez-Sanchez EP, Cox DL, Cosby D & Gomez-Sanchez CE 1995 Cloning, expression, and tissue distribution of the rat nicotinamide adenine dinucleotide-dependent 11 beta-hydroxysteroid dehydrogenase. Endocrinology 136 3729–3734.[Abstract]
Received 5 October 2004
Accepted 11 January 2005
Made available online as an Accepted Preprint 4 February 2005
This article has been cited by other articles:
 |
I. J.N. Padmavathi, K. R. Rao, L. Venu, M. Ganeshan, K. A. Kumar, Ch. N. Rao, N. Harishankar, A. Ismail, and M. Raghunath Chronic Maternal Dietary Chromium Restriction Modulates Visceral Adiposity: Probable Underlying Mechanisms Diabetes, January 1, 2010; 59(1): 98 - 104. [Abstract] [Full Text] [PDF] |
||||
|
|
H. Zhou, W. Mak, R. Kalak, J. Street, C. Fong-Yee, Y. Zheng, C. R. Dunstan, and M. J. Seibel Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice Development, February 1, 2009; 136(3): 427 - 436. [Abstract] [Full Text] [PDF] |
||||
|
|
Y. Liu, D. Mladinov, J. L. Pietrusz, K. Usa, and M. Liang Glucocorticoid response elements and 11{beta}-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression Cardiovasc Res, January 1, 2009; 81(1): 140 - 147. [Abstract] [Full Text] [PDF] |
||||
|
|
R. J Webb, N. Sunak, L. Wren, and A. E Michael Inactivation of glucocorticoids by 11{beta}-hydroxysteroid dehydrogenase enzymes increases during the meiotic maturation of porcine oocytes Reproduction, December 1, 2008; 136(6): 725 - 732. [Abstract] [Full Text] [PDF] |
||||
|
|
N. Schliebe, R. Strotmann, K. Busse, D. Mitschke, H. Biebermann, L. Schomburg, J. Kohrle, J. Bar, H. Rompler, J. Wess, et al. V2 vasopressin receptor deficiency causes changes in expression and function of renal and hypothalamic components involved in electrolyte and water homeostasis Am J Physiol Renal Physiol, October 1, 2008; 295(4): F1177 - F1190. [Abstract] [Full Text] [PDF] |
||||
 |
S. Senesi, P. Marcolongo, I. Manini, R. Fulceri, V. Sorrentino, M. Csala, G. Banhegyi, and A. Benedetti Constant expression of hexose-6-phosphate dehydrogenase during differentiation of human adipose-derived mesenchymal stem cells J. Mol. Endocrinol., September 1, 2008; 41(3): 125 - 133. [Abstract] [Full Text] [PDF] |
||||
|
|
A. E. Michael and A. T. Papageorghiou Potential significance of physiological and pharmacological glucocorticoids in early pregnancy Hum. Reprod. Update, September 1, 2008; 14(5): 497 - 517. [Abstract] [Full Text] [PDF] |
||||
|
|
F. Jeanneteau, M. J. Garabedian, and M. V. Chao Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect PNAS, March 25, 2008; 105(12): 4862 - 4867. [Abstract] [Full Text] [PDF] |
||||
 |
K S Fegan, M T Rae, H O D Critchley, and S G Hillier Anti-inflammatory steroid signalling in the human peritoneum J. Endocrinol., February 1, 2008; 196(2): 369 - 376. [Abstract] [Full Text] [PDF] |
||||
|
|
H. Zhou, W. Mak, Y. Zheng, C. R. Dunstan, and M. J. Seibel Osteoblasts Directly Control Lineage Commitment of Mesenchymal Progenitor Cells through Wnt Signaling J. Biol. Chem., January 25, 2008; 283(4): 1936 - 1945. [Abstract] [Full Text] [PDF] |
||||
|
|
C. E. Perrone, D. A. L. Mattocks, G. Hristopoulos, J. D. Plummer, R. A. Krajcik, and N. Orentreich Methionine restriction effects on 11 -HSD1 activity and lipogenic/lipolytic balance in F344 rat adipose tissue J. Lipid Res., January 1, 2008; 49(1): 12 - 23. [Abstract] [Full Text] [PDF] |
||||
|
|
W. C. Cooper, Y. Jin, and T. M. Penning Elucidation of a Complete Kinetic Mechanism for a Mammalian Hydroxysteroid Dehydrogenase (HSD) and Identification of All Enzyme Forms on the Reaction Coordinate: THE EXAMPLE OF RAT LIVER 3{alpha}-HSD (AKR1C9) J. Biol. Chem., November 16, 2007; 282(46): 33484 - 33493. [Abstract] [Full Text] [PDF] |
||||
|
|
Z. Yang, C. Guo, P. Zhu, W. Li, L. Myatt, and K. Sun Role of glucocorticoid receptor and CCAAT/enhancer-binding protein {alpha} in the feed-forward induction of 11{beta}-hydroxysteroid dehydrogenase type 1 expression by cortisol in human amnion fibroblasts J. Endocrinol., November 1, 2007; 195(2): 241 - 253. [Abstract] [Full Text] [PDF] |
||||
|
|
S. G Hillier Diamonds are forever: the cortisone legacy J. Endocrinol., October 1, 2007; 195(1): 1 - 6. [Abstract] [Full Text] [PDF] |
||||
|
|
D. Rogoff, J. W. Ryder, K. Black, Z. Yan, S. C. Burgess, D. R. McMillan, and P. C. White Abnormalities of Glucose Homeostasis and the Hypothalamic-Pituitary-Adrenal Axis in Mice Lacking Hexose-6-Phosphate Dehydrogenase Endocrinology, October 1, 2007; 148(10): 5072 - 5080. [Abstract] [Full Text] [PDF] |
||||
|
|
C Chandras, T E Harris, A L. Bernal, D R E Abayasekara, and A E Michael PTGER1 and PTGER2 receptors mediate regulation of progesterone synthesis and type 1 11{beta}-hydroxysteroid dehydrogenase activity by prostaglandin E2 in human granulosa lutein cells J. Endocrinol., September 1, 2007; 194(3): 595 - 602. [Abstract] [Full Text] [PDF] |
||||
|
|
C. S. Wyrwoll, P. J. Mark, and B. J. Waddell Developmental Programming of Renal Glucocorticoid Sensitivity and the Renin-Angiotensin System Hypertension, September 1, 2007; 50(3): 579 - 584. [Abstract] [Full Text] [PDF] |
||||
|
|
C. Jang, V. R. Obeyesekere, R. J. Dilley, Z. Krozowski, W. J. Inder, and F. P. Alford Altered Activity of 11{beta}-Hydroxysteroid Dehydrogenase Types 1 and 2 in Skeletal Muscle Confers Metabolic Protection in Subjects with Type 2 Diabetes J. Clin. Endocrinol. Metab., August 1, 2007; 92(8): 3314 - 3320. [Abstract] [Full Text] [PDF] |
||||
|
|
K. Usa, R. J. Singh, B. C. Netzel, Y. Liu, H. Raff, and M. Liang Renal interstitial corticosterone and 11-dehydrocorticosterone in conscious rats Am J Physiol Renal Physiol, July 1, 2007; 293(1): F186 - F192. [Abstract] [Full Text] [PDF] |
||||
|
|
L M Thurston, D R E Abayasekara, and A E Michael 11{beta}-Hydroxysteroid dehydrogenase expression and activities in bovine granulosa cells and corpora lutea implicate corticosteroids in bovine ovarian physiology J. Endocrinol., May 1, 2007; 193(2): 299 - 310. [Abstract] [Full Text] [PDF] |
||||
|
|
P. Marcolongo, S. Piccirella, S. Senesi, L. Wunderlich, I. Gerin, J. Mandl, R. Fulceri, G. Banhegyi, and A. Benedetti The Glucose-6-Phosphate Transporter-Hexose-6-Phosphate Dehydrogenase-11{beta}-Hydroxysteroid Dehydrogenase Type 1 System of the Adipose Tissue Endocrinology, May 1, 2007; 148(5): 2487 - 2495. [Abstract] [Full Text] [PDF] |
||||
|
|
G. Groyer, B. Eychenne, C. Girard, K. Rajkowski, M. Schumacher, and F. Cadepond Expression and Functional State of the Corticosteroid Receptors and 11{beta}-Hydroxysteroid Dehydrogenase Type 2 in Schwann Cells Endocrinology, September 1, 2006; 147(9): 4339 - 4350. [Abstract] [Full Text] [PDF] |
||||
|
|
C. Bruley, V. Lyons, A. G. F. Worsley, M. D. Wilde, G. D. Darlington, N. M. Morton, J. R. Seckl, and K. E. Chapman A Novel Promoter for the 11{beta}-Hydroxysteroid Dehydrogenase Type 1 Gene Is Active in Lung and Is C/EBP{alpha} Independent Endocrinology, June 1, 2006; 147(6): 2879 - 2885. [Abstract] [Full Text] [PDF] |
||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HOME | HELP | CONTACT US | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |