The new biology of aldosterone
- MRC Blood Pressure Group, Division of Cardiovascular and Medical Sciences, University of Glasgow, Gardiner Institute, Western Infirmary, Glasgow G11 6NT, UK
- (Requests for offprints should be addressed to J M C Connell; Email: j.connell{at}clinmed.gla.ac.uk)
Abstract
Classically, aldosterone is synthesised in the adrenal zona glomerulosa and binds to specific mineralocorticoid receptors located in the cytosol of target epithelial cells. Translocation of the resulting steroid receptor complex to the cell nucleus modulates gene expression and translation of specific ‘aldosterone-induced’ proteins that regulate electrolyte and fluid balance. However, non-epithelial and rapid non-genomic actions of aldosterone have also been described that account for a variety of actions of aldosterone that contribute to blood pressure homeostasis. These include key actions on endothelial cells and on cardiac tissue.
There is also evidence that aldosterone can be synthesised in other tissues; the most convincing evidence relates to the central nervous system. However, suggestions that aldosterone is produced in the heart remain controversial, and adrenal derived aldosterone is the principal source of circulating and locally available hormone.
Recent studies have shown major therapeutic benefits of mineralocorticoid receptor antagonism in cardiac failure, which emphasise the importance of aldosterone in causing adverse cardiovascular pathophysiological effects. Additional evidence demonstrates that aldosterone levels predict development of high blood pressure in normotensive subjects, while it is now clear that increased aldosterone action contributes to hypertension and cardiovascular damage in approximately 10% of patients with established hypertension.
These new findings highlight the role of aldosterone as a key cardiovascular hormone and extend our understanding of its role in determining adverse cardiovascular outcomes.
Introduction
It is now over 50 years since Sylvia and James Tait, in collaboration with Tadeusz Reichstein, isolated and characterized the steroid hormone, aldosterone (Tait et al. 2004). Since then, the traditional view has been that the hormone is synthesized in the adrenal gland and binds to specific mineralocorticoid receptors (MRs) located in the cytosol of target epithelial cells. This steroid receptor complex then translocates to the cell nucleus where it modulates gene expression and translation of specific ‘aldosterone-induced’ proteins that regulate electrolye and fluid balance and subsequent blood-pressure homoeostasis. However, a number of recent studies show that aldosterone also has marked effects in a wide range of non-epithelial tissues (such as the heart), that it can be synthesized and regulated in a number of extra-adrenal tissues and that it may act through alternative receptors in epithelial and non-epithelial tissue in a rapid non-genomic manner which is independent of gene transcription and translation.
Recently, there has been a new wave of interest in aldosterone due mainly to its growing profile as a key cardiovascular hormone. The importance of aldosterone in cardiovascular homoeostasis is evident in animal models where expression of components of the aldosterone biosynthetic pathway or of mineralocorticoid action is abolished or increased, and in the rare human monogenic syndromes that result in either an absence or an excess of aldosterone. Recent work now shows that aldosterone levels within the upper part of the physiological range predispose normotensive subjects to the development of hypertension and that major therapeutic benefits result from MR antagonism in cardiac failure (Vasan et al. 2004).
In this review we outline some of the recent advances in our knowledge of aldosterone biosynthesis, its novel sites of action and the mechanism and specificity of this action. In a relatively brief article it is not possible to focus in detail on all aspects of this topic, and we have referred to key reviews where possible. Some aspects (e.g. the role of the 11β-hydroxysteroid dehydrogenase (11β-HSD) enzyme to maintain MR specificity and the rapid non-genomic actions of steroids) will be covered in greater detail elsewhere in this series of reviews.
Current concepts
Aldosterone biosynthesis
Aldosterone is synthesized from cholesterol in the zona glomerulosa (ZG) of the adrenal cortex by a series of locus- and orientation-specific enzymatic reactions (Fig. 1⇓). Firstly, cholesterol must cross the outer mitochondrial membrane to the inner membrane where the first enzyme in the steroidogenic pathway is located. A number of cholesterol-carrier/-translocation systems have been identified including sterol carrier protein and peripheral benzodiazepine receptors. However, the process is now known to be mediated by steroidogenic acute regulatory protein (StAR), which forms a core through the membrane. StAR is present in all steroidogenic tissue (Stocco 2001a) and evidence that it plays a key role in steroidogeneis came from studies of patients with congenital lipoid adrenal hyperplasia, whereby mutations of StAR gene resulted in an inability to make steroids and accumulation of cholesterol in the adrenal glands (Lin et al. 1995). Similarly, the StAR-knockout mouse has elevated lipid deposits in the adrenal cortex and extremely low levels of steroid despite elevated levels of adrenocorticotrophic hormone (corticotrophin; ACTH) and cortocotrophin-releasing hormone (CRH; Caron et al. 1997). StAR gene expression is increased by most agents know to stimulate steroid biosynthesis and the translocation of cholesterol to the inner mitochondrial matrix is now considered to be the rate-limiting step in steroidogenesis (Stocco 2001b).
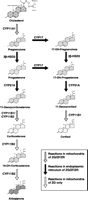
The biosynthesis of aldosterone and cortisol in the human adrenal cortex. ZG, zona glomerulosa; ZF, zona fasciculata; ZR, zona reticularis.
Following translocation to the mitochondrion, cholesterol is converted to aldosterone by a series of enzymatic reactions catalysed by dehydrogenases and mixed-function oxidases, many of which belong to the cytochrome P450 (CYP) superfamily of haem-containing enzymes. These require a coupled coenzyme system (adrenodoxin/adrenodoxin reductase) which transfers electrons to the P450 enzyme, acting as reducing equivalents for the hydroxylation reaction (Lambeth et al. 1982). The first reaction is the conversion of cholesterol to pregnenolone, catalysed by the P450 side-chain cleavage enzyme (CYP11A1), encoded by the CYP11A1 gene on human chromosome 15 (Fig. 1⇑). This enzyme catalyses three reactions, which before the discovery of StAR were considered to be rate-limiting: a 20α-hydroxylation, a 22-hydroxylation and cleavage of the bond between C-20 and C-22 to produce pregnenolone and isocaproic acid (Lieberman & Lin 2001). Pregnenolone is released into the cytosol and is converted to progesterone by dehydrogenation of the 3β-hydroxyl group and isomerization of the double bond at C-5 to Δ4 by 3β-hydroxysteroid dehydrogenase (3β-HSD), which is located on the membrane of the smooth endoplasmic reticulum. Two 3β-HSD isoenzymes have been identified in humans and several in the rat and mouse (Mason 1993, Simard et al. 1993). HSD3B1 and HSD3B2 are encoded by the 3β-HSD1 and 3β-HSD2 genes, respectively, which lie in tandem on chromosome 1p13·1 (Berube et al. 1989, Lachance et al. 1991).
Progesterone undergoes 21-hydroxylation by the CYP21A enzyme located on the cytoplasmic surface of the smooth endoplasmic reticulum, producing 11-deoxycorticosterone (DOC; Shinzawa et al. 1988). The gene (CYP21A) encoding this enzyme was mapped to human chromosome 6p21·3 and is adjacent to a pseudogene, CYP21P (White et al. 1986). The conversion of DOC to aldosterone involves three consecutive reactions: 11β-hydroxylation of DOC to form corticosterone, 18-hydroxylation to yield 18-hydroxycorticosterone (18-OH-B) and finally 18-methyloxidation to aldosterone. These reactions are catalysed by aldosterone synthase, which is located on the inner mitochondrial membrane and encoded by the CYP11B2 gene, which is expressed in the ZG. Aldosterone synthase is highly homologous (approximately 93%) to 11β-hydroxylase (the product of the CYP11B1 gene), which catalyses the conversion of 11-deoxycortisol to the glucocorticoid cortisol. The two genes are located in tandem on human chromosome 8q21–22 (Chua et al. 1987, Mornet et al. 1989, Wagner et al. 1991). Only two CYP11B genes are found in humans but rats possess a CYP11B3 gene and a CYP11B4 gene. CYP11B4 appears to be a pseudogene, but CYP11B3 encodes an enzyme possessing 18- and 11-hydroxylase activities which is expressed in the adrenal zona fasciculata-reticularis for a short period after birth and is regulated by ACTH (Mellon et al. 1995, Zhou et al. 1995).
Regulation of aldosterone biosynthesis
A number of factors have been shown to stimulate or inhibit aldosterone production, including adrenaline, vasoactive intestinal polypeptide, serotonin, ouabain, atrial natriuretic peptide, dopamine, heparin and adrenomedullin. More recently, novel factors secreted by adipose tissue have also been shown to stimulate aldosterone synthesis in vitro. However, the principal regulators of aldosterone synthesis and secretion of aldosterone are angiotensin II (Ang II), the concentration of extracellular potassium and ACTH (Muller 1987, Quinn & Williams 1988).
Renin–angiotensin system (RAS)
Aldosterone biosynthesis is regulated principally by the RAS. Renin is synthesized and released by the juxtaglomerular cells in the afferent arteriole of the kidney in response to a decrease in intravascular volume detected by baroreceptors (mediated by β-adrenoreceptor activation) and by a reduced sodium concentration at the macula densa. Renin catalyses the hydrolysis of angiotensinogen to angiotensin I (Ang I) which is then converted to Ang II by angiotensin-converting enzyme (ACE), present in the lungs and vascular tissue. Complete RASs have also been described in other tissues, including the brain, vasculature and adrenal cortex (Samani 1994, MacKenzie et al. 2002a).
Ang II acts on vascular smooth muscle to cause vaso-constriction, and on the adrenal ZG to stimulate aldosterone production. The adrenal response to Ang II occurs within minutes, a time course that implies that no new protein synthesis is required. This acute, Ang II-mediated release of aldosterone may involve rapid synthesis from intermediate compounds in the steroidogenic pathway or de novo synthesis from cholesterol, possibly as a consequence of StAR protein activation, leading to increased transport of cholesterol to the inner mitochondrial membrane. Chronic stimulation by Ang II results in ZG hypertrophy and hyperplasia, increased CYP11B2 expression and subsequent aldosterone secretion.
The mechanism by which Ang II stimulates aldosterone production is not yet fully understood. However, it is known to act on specific G-protein-coupled receptors (AT1 receptors) which cause phospholipase C to stimulate intracellular production of inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG), which then activate protein kinase C (PKC). IP3 also increases the concentration of intracellular free calcium ([Ca2+]i), causing several Ca2+/calmodulin-dependent protein kinases (CaM kinases) to phosphorylate and activate such transcription factors as activating transcription factor (ATF)-1, ATF-2 and cAMP-response-element (CRE)-binding protein (CREB; Spat & Hunyady 2004). These bind CRE and other cis-acting elements (e.g. Ad-5 and NBRE-1) which are unique to the 5′ untranslated region (5′UTR) of the CYP11B2 gene. The NBRE-1 and Ad-5 cis-elements are specific to CYP11B2 and mutation of these sites is known to decrease gene expression in reporter constructs. The Ad-5 cis-element binds members of the NGFIB family as well as steroidogenic factor 1 (SF-1) and chicken ovalbumin upstream promoter-transcription factor (COUP-TF). Recent studies have identified the transcription factor NURR-1 as a key regulator of CYP11B2 transcription that responds to Ang II; its expression is upregulated in aldosterone-secreting tumours (Bassett et al. 2004).
Potassium ions (K+)
Extracellular K+ concentration is the other key determinant of aldosterone secretion. Indeed, production of aldosterone is acutely sensitive to very small changes in extracellular [K+]. Increased [K+] concentration stimulates aldosterone secretion, helping to maintain K+ homoeostasis. The effects of extracellular [K+] and Ang II are synergistic, so that the prevailing [K+] determines the concentration/effect relationship for Ang II-mediated aldosterone production (Spat 2004).
Increased extracellular [K+] causes ZG cell-membrane depolarization, leading to the opening of voltage-dependent L- and T-type Ca2+ channels and a rapid rise in [Ca2+]i.This leads to the activation of calmodulin and CaM kinases which phosphorylate transcription factors to stimulate CYP11B2 gene transcription, as mentioned previously (Spat & Hunyady 2004). Ang II and K+ therefore regulate CYP11B2 transcription through common Ca2+-dependent signaling pathways and also through common transcription factors (Clyne et al. 1997).
ACTH
The hypothalamic/pituitary adrenal axis primarily controls glucocorticoid production. However, its effector peptide, ACTH, also contributes to the regulation of aldosterone biosynthesis. ACTH interacts with specific receptors in the adrenal cortex to stimulate production of glucocorticoids. It also stimulates adrenal blood flow and chronic increases results in adrenal hyperplasia and hyptrophy of the adrenal zona fasciculata (Quinn & Williams 1988). Acutely, ACTH stimulates aldosterone production via cAMP-mediated pathways and protein-synthesis-independent mechanisms involving macrophage-derived factor, steroidogenesis-inducing protein and calmidazolium (Cozza et al. 1990, Cooke 1999). In contrast, chronic excess of ACTH suppresses plasma aldosterone levels in both humans and animal models (Fuchs-Hammoser et al. 1980, Holland & Carr 1993, Aguilera et al. 1996). The mechanism of chronic inhibition is unclear but cAMP may downregulate the expression of Ang II receptors in adrenocortical cells (Yoshida et al. 1991, Bird et al. 1994). Alternatively, ACTH may transform proliferating ZG cells into zona fasciculata cells or divert precursors from the mineralocorticoid to the glucocorticoid pathway (McAllister & Hornsby 1988, Bird et al. 1996, Vinson 2003).
Despite the opposing effects of acute and chronic ACTH, there is no doubt that the hormone is involved with normal physiological regulation of aldosterone production. For example, aldosterone secretion displays a diurnal variation with higher levels in the morning and lower levels later in the day; this is entrained by ACTH (Richards et al. 1986). Furthermore, recent evidence shows that the pro-opiomelanocortin (POMC)-knockout mouse has abnormal adrenocortical morphology and has reduced (but detectable) levels of aldosterone, suggesting that ACTH is required for normal aldosterone secretion (Coll et al. 2004).
Adipose tissue factors
Earlier studies suggested that lipid-derived factors can acutely stimulate aldosterone synthesis (Goodfriend et al. 2002). A variety of fatty acid molecules have been proposed as candidates and there is evidence to suggest that their aldosterone-stimulating properties are dependent on their oxidation by the liver (Goodfriend et al. 1998). More recently, in vitro experiments identified a high-molecular-weight protein released by adipose cells that can stimulate synthesis of aldosterone by adrenocortical cells; the physiological relevance and importance of this is not clear (Ehrhart-Bornstein et al. 2003). However, it is known that increased aldosterone production is a feature of adipose-associated hypertension in dogs and may therefore be a consequence of factors released by omental fat (de Paula et al. 2004). However, increased visceral adiposity is also associated with hyper-insulinaemia; insulin is known to increase aldosterone synthesis in vitro and also to increase renal sodium reabsorption, possibly by activating the phosphoinositide 3-kinase (PI3K)/serum- and glucocorticoid-regulated kinase-1 (Sgk1) pathway in synergy with aldosterone (Pearce 2003, Goodfriend & Calhoun 2004). So, the precise contribution made to aldosterone synthesis, and to the sodium retention caused by aldosterone, by the range of factors derived from adipose tissue or associated with obesity is still to be determined accurately.
Epithelial actions of aldosterone
Classically, aldosterone acts on epithelial cells, particularly in the renal collecting duct, but also in the parotid gland and colon, where it regulates the transport of Na+, K+ and water (Fig. 2⇓). Aldosterone-responsive epithelial cell monolayers act as barriers separating the internal and external environment; they also permit the reabsorption of Na+ and water. These functions are facilitated by the lipid composition of the apical membrane and by the formation of high-resistance tight junctions. Transport through these cells is facilitated by an electrochemical potential across the apical membrane from urine to cell and by an active-transport mechanism across the basolateral membrane from cell to interstitium. Sodium reabsorption across the apical membrane is mediated by the luminal amiloride-sensitive epithelial sodium Na+ channel (ENaC). Transport across the basolateral membrane is driven by the ouabain-sensitive serosal Na+/K+-ATPase which drives the entry of sodium and the excretion of potassium from the cell to the lumen through the luminal K+ channel. Water follows the movement of Na+ across the monolayer. These are considered to be the principal mediators of aldosterone action in epithelial cells. However, other protein targets in the apical membrane have also been identified – e.g. the luminal Na+/H+ exchanger (NHE3) in the colon and the luminal thiazide-sensitive Na+/Cl− cotransporter in the distal renal tubule – which appear to mediate sodium reabsorption in response to volume depletion (Cho et al. 1998, Kim et al. 1998). While the overall action of aldosterone on electrolyte transport is clear, the exact mechanism by which it exerts these effects is unknown. Apical channel activity is the limiting step in the transport process, and it is likely that aldosterone ultimately acts to increase the open time of the existing ion channels or by increasing the total number of channels; current evidence suggests that it can do both.
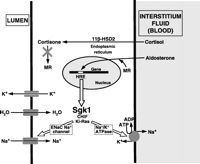
The classical genomic action of aldosterone on epithelial tissue. HRE, hormone response element.
Mechanism of action of aldosterone
The effects of aldosterone are mediated by receptors located in the cell cytosol. These belong to the nuclear receptor superfamily and are composed of several functional domains which include an N-terminal domain, a highly-conserved DNA-binding domain and a C-terminal ligand-binding domain (Arriza et al. 1987). They exist in the cytosol as a hetero-oligomeric complex with heat-shock proteins and immunophilins. Hormone binding results in a conformational change resulting in dissociation of the associated proteins, dimerization and translocation to the cell nucleus (Rogerson et al. 2004). The activated receptor/hormone complex binds to steroid responsive elements in the 5′UTR of aldosterone-responsive genes that activate or repress gene transcription. The receptor–steroid complex may also act through a process known as transcription interference or synergy (Karin 1998), whereby it interacts with other transcription factors that themselves bind DNA to activate or repress transcriptional activity. This enables transcriptional modulation without direct interaction between the receptor–steroid complex and DNA. The epithelial action of aldosterone consists of early (1–6 h) and late (>6 h) phases. The early phase is mediated by changes in gene expression that activate ion channels and signalling proteins which then induce electrolyte-transport proteins. The late phase results from both primary and secondary effects on gene expression. Of the induced genes and proteins, Sgk1 is most firmly established as a mediator of aldosterone action, although there are other candidates. These are detailed below.
Sgk1
One of the early aldosterone-induced proteins that has received considerable attention is Sgk1, a serine-threonine kinase (Stockand 2002, Naray-Fejes-Toth et al. 2004). Studies in Xenopus laevis oocytes have shown that overexpression of Sgk1 together with ENaC activates the sodium channel; it also appears to activate the Na+/K+-ATPase and Na+/K+/Cl− cotransporter. Recent studies in intact mice confirmed that aldosterone causes a marked rise in the expression of Sgk1 in the distal tubule of the kidney. The importance of this molecule in aldosterone action is further illustrated by studies of a transgenic mouse model whose Sgk1 lacks a functional kinase. These animals have mild pseudohypoaldosteronism, but sodium restriction promotes Na+ wasting and hyperkalemia (Wulff et al. 2002). The mechanism by which Sgk-1 acts remains unknown but it has been suggested that it binds and phosphorylates ENaC regulatory protein, also known as neuronal precursor cells-expressed developmentally down-regulated protein 4–2 (Nedd4–2), to reduce its binding to ENaC (Synder et al. 2002). A subsequent reduction in ENaC ubiquitination by Nedd4–2 increases ENaC density and stability at the apical membrane resulting in increased ENaC-dependent Na+ reabsorption.
Kirsten Ras GTP-binding protein-2A (Ki-RasA)
The expression of the small, monomeric Ki-RasA is induced during the early phase of aldosterone action and appears to be necessary for the effects of aldosterone on sodium transport in renal epithelial cells. Ki-RasA appears to have dual contrasting effects on the ENaC channel. Overexpression in X. laevis oocytes showed that Ki-RasA not only keeps the channel open but also decreases the number of channels in the plasma membrane (Mastroberardino et al. 1998, Stockand 2002, Stockand & Meszaros 2003). The mechanism by which it modulates these action remains unclear.
PI3K
The lipid kinase, PI3K, is thought to play a role in the actions of aldosterone, insulin and vasopressin. While it is not considered an aldosterone-induced protein, its activity is increased by aldosterone and insulin in the kidney (Blazer-Yost et al. 1998, Edinger et al. 1999, Stockand 2002). Inhibition of PI3K reduces both the early and late actions of aldosterone. Similar effects are observed on insulin-induced Na+ transport; stimulation of sodium transport in the kidney by antidiuretic hormone is also partly dependent on PI3K. Therefore, this kinase may represent a central point where the signalling pathways of aldosterone, insulin and vasopressin integrate and activate ENaC and Na+/K+-ATPase by a collective pathway.
Corticosteroid hormone-induced factor (CHIF)
CHIF is expressed in the basolateral membranes of epithelial cells in the distal colon and nephron. It belongs to the FXYD transmembrane family of seven proteins which contain both this motif and three other conserved amino acids which serve to regulate ion channels and transport proteins (the γ-subunit of Na+/K+-ATPase also belongs to this family; Stockand 2002, Geering et al. 2003). Some studies have shown that CHIF, FXYD7 and FXYD1 associate specifically with Na+/K+-ATPase, suggesting that members of this protein group may substitute for one another (Farman et al. 2003). The mechanism of signal transduc-tion is unknown. Presumably, aldosterone stimulates CHIF expression and the resulting protein interacts with final effectors to promote transport. The role of CHIF in electrolyte balance is highlighted in studies of CHIF-knockout mice; potassium loading plus treatment with furosemide was lethal in the knockout but not the wild-type controls (Aizman et al. 2002, Garty et al. 2003).
Target tissue specificity
Until the 1980s, the specificity of aldosterone action was believed to be conferred by the presence of high-affinity type 1 MRs, which were first described in the renal cytosol (Arriza et al. 1987). These are abundant in aldosterone target tissues including the kidney, colon and salivary glands; they are distinguishable from the type 2 glucocorticoid receptors (GRs), which are expressed ubiquitously and exhibit a higher affinity for glucocorticoids such as cortisol and corticosterone. However, further studies of the MR in cultured pituitary cells demonstrated that it also had an equally high affinity for glucocorticoids, reflected in the high degree of homology between MRs and GRs in their DNA-binding domains (94%) and ligand-binding domains (57%). The lack of specificity of the MR and the fact that plasma levels of glucocorticoids are 1000 times greater than those of aldosterone suggests that MRs should be predominantly occupied by glucocorticoid. Aldosterone’s ability to act on MRs was partly explained by the fact that the majority of glucocorticoids are bound to proteins in plasma. However, this still left a 100-fold concentration difference in glucocorticoids’ favour, so it was clear that an additional mechanism must also exist.
The key explanation was provided by seminal studies which characterized the 11β-HSD type 2 (11β-HSD2) enzyme complex, which colocalizes with MRs in target epithelial tissues and is also found in placenta and vascular tissue (Funder et al. 1988, Stewart et al. 1988). This catalyses the conversion of active glucocorticoids, capable of binding with high affinity to MRs, into inactive metabolites (in humans, this is the conversion of cortisol to cortisone). These metabolites have little affinity for the MR and so the action of 11β-HSD2 effectively protects the MR from illicit occupation by glucocorticoids. The importance of this mechanism for maintaining the specificity of the MR for occupation by aldosterone is confirmed by the syndrome of apparent mineralocorticoid excess, characterized by sodium retention, hypokalemia low renin and hypertension, in the absence of excessive aldosterone or DOC. This syndrome can be caused by excessive consumption of liquorice, a component of which – glycerrhetinic acid – inhibits 11β-HSD2 (Stewart et al. 1988). A more severe form of the syndrome, inherited in an autosomal recessive manner, results from mutations to the gene encoding 11β-HSD2 that inactivate the resulting protein. Excessive MR activation caused by cortisol in this circumstance can be treated with the synthetic glucocorticoid dexamethasone, which suppresses cortisol secretion but does not bind MRs (Stewart et al. 1996, White et al. 1997, Stewart & Krozowski 1999).
Although this mechanism accounts for aldosterone specificity in epithelia, it fails to explain how aldosterone can act in cells and tissues which possess abundant MRs but no or little 11β-HSD2, such as cardiac fibroblasts (see below). In addition, the efficiency of 11β-HSD2 alone may not be sufficient to account for the complete lack of MR activation by glucocorticoids. However, molecular studies of the cloned MR show that even though mineralocorticoids and glucocorticoids bind with equivalent affinity, aldosterone achieves greater transactivation. This suggests that ligands can have differential effects on gene transcription at the same receptor. These may be due to conformational differences in the MR–ligand complex, leading to variable degrees of stability. The interaction with co-activators after binding to response elements may also be dependent on the ligand (Farman & Rafestin-Oblin 2001). Finally, very recent studies suggest that high local concentrations of NADH, which is produced by 11β-HSD2, inactivates MR–glucocorticoid complexes, providing another potential mechanism to maintain MR specificity (Funder 2004).
Non-epithelial actions of aldosterone
MRs have also been localized in a number of non-epithelial tissues, particularly in the cardiovascular system and central nervous system (CNS). While the properties of the MRs in these tissues are largely similar, the effects they mediate are extremely diverse. In contrast to its established effects on electrolyte balance in epithelial tissue, aldosterone in the cardiovascular system promotes cardiac hypertrophy, fibrosis and abnormal vascular endothelial function. In the CNS, it appears to regulate blood pressure, salt appetite and sympathetic tone.
Cardiovascular system
Excess aldosterone secretion results in hypertension, which may be due in part to its direct actions on the cardiovascular system. In the vascular system, aldosterone is known to modulate vascular tone, possibly by increasing the pressor response to catecholamines and impairing the vasodilatory response to acetylcholine or by upregulation of Ang II receptors (Schiffrin et al. 1985, Jazayeri & Meyer 1989, Wang et al. 1992, Taddei et al. 1993). This latter effect is consistent with reduced availability of endothelial nitric oxide. In addition, aldosterone excess also promotes collagen deposition in blood vessels, enhancing vascular remodelling at the expense of compliance. This remodelling was originally regarded as an adaptation to the arterial hypertension caused by aldosterone excess. However, chronic aldosterone administration in the presence of NaCl has been shown to stimulate perivascular and interstitial cardiac fibrosis and cardiac hypertrophy independently of changes in blood pressure (Brilla & Weber 1992, Funder 1995, Rocha & Stier 2001). Indeed, left-ventricular hypertrophy and stroke are more common in age- and sex-matched patients with primary aldosteronism than in essential hypertension of similar severity (Tanabe et al. 1997, Rossi et al. 1997). Interestingly, treatment of severe heart failure patients with the MR antagonists spironolactone and eplerenone, at doses which do not significantly alter blood pressure, significantly reduces the risk of both morbidity and mortality (Pitt et al. 1999, 2003b). In addition, recent reports suggest that eplerenone can not only prevent but also reverse mineralocorticoid induced fibrosis in animal models (Young & Funder 2004).
The mechanism of aldosterone-induced fibrosis is unclear, but aldosterone has been shown to increase collagen I synthesis in cardiac fibroblasts (Robert et al. 1994). It may also increase endothelin receptor numbers which increases collagen synthesis (Fullerton & Funder 1994). The deleterious effects of aldosterone in animal models are also dependent on a high sodium intake. The way in which this exacerbates the effects of aldosterone on the heart are not fully understood, but it does not appear to be directly related to blood pressure. Histological features of aldosterone-induced cardiac fibrosis include proliferation of cardiac myocytes and fibroblasts and intense perivascular inflammation. Most pathophysiological cardiac effects of aldosterone have a time course of at least several weeks and are therefore probably MR-dependent, although some inflammatory markers (tumour necrosis factor α and ED-1) and increases in collagen III deposition have been observed as early as day 2 in some models of mineralocorticoid-induced fibrosis (Young et al. 2003). Specific MRs have been detected in cardiac myocytes, endocardium, vascular smooth muscle and endothelial cells and 11β-HSD2 expression has been demonstrated in human and rodent heart tissue, mainly in cardiac blood vessels (Lombes et al. 1995, Kayes-Wandover & White 2000). Overexpression of MR in the mouse heart results in left-ventricular hypertrophy with normal blood pressure, consistent with the idea that cardiac hypertrophy is caused by direct effects of aldosterone on heart rather than indirectly through high blood pressure. 11β-HSD2 expression has been demonstrated in the heart by some, but not all, groups (Lombes et al. 1995, Slight et al. 1996, Kayes-Wandover & White 2000, Sheppard & Autelitano 2002). However, the levels of expression are generally low compared with those found in epithelial tissue and may be limited to specific cell types, e.g vascular smooth muscle cells.
It has been proposed that cardiac biosynthesis of aldosterone (see below) could result in concentrations high enough to occupy MRs without the need for glucocorticoid inactivation, although several studies suggest that this is unlikely (Silvestre et al. 1998a, Gomez-Sanchez et al. 2004). Further evidence against a role for local aldosterone comes from animal models of fibrosis induced by Ang II and Na+, an effect which is abolished completely by adrenalectomy (Rocha et al. 2002), suggesting the effects are mediated or permitted by a factor derived from the adrenal gland.
In addition, glucocorticoids may also interact with cardiac MRs to influence the net effects of aldosterone. Under normal circumstances, it is likely that cardiac MRs are occupied by glucocorticoid due to its higher circulating concentration. This is also likely to be augmented by the reductase activity (converting cortisone to cortisol) of the type 1 11β-HSD enzyme (11βHSD1), observed in rat cardiac fibroblasts (Sheppard & Autelitano 2002). However, concomitant infusion of corticosterone and aldosterone in the rat significantly attenuates the rise in blood pressure and the cardiac fibrosis caused by aldosterone alone, suggesting that corticosterone is a MR antagonist in cardiomyocytes (Young & Funder 2000). Similarly, transgenic mice overexpressing 11β-HSD2 in cardiomyocytes developed cardiac hypertrophy, fibrosis and heart failure on a normal-salt diet. Eplerenone treatment reversed these effects. These studies confirm the detrimental effects of cardiac mineralocorticoid activation by aldosterone and highlight the protective role of glucocorticoids in preventing the harmful actions of aldosterone (Qin et al. 2003).
CNS
Aldosterone has also been shown to exert a number of effects in the CNS (de Kloet et al. 1987, Gomez-Sanchez 1997). Facilitated by its lipophilic properties, exogenous aldosterone can cross the blood–brain barrier relatively easily, although much of it may be pumped back across the barrier by the multidrug-resistant P-glycoprotein (Ueda et al. 1992, Karssen et al. 2001). Compared with GRs, which are distributed widely throughout the CNS in neurons and glial cells, there are fewer MRs and these are concentrated predominantly in the hippocampus, the septum and the granular cells of the cerebellum (Reul & de Kloet 1986, Agarwal et al. 1993). In contrast to epithelial tissue, MRs in the CNS do not appear to co-localize with 11β-HSD2 (Diaz et al. 1998). High 11β-HSD2 levels in the adult rat brain are restricted to the subcommissural organ (SCO), with lower levels in the hypothalamic ventromedial nucleus and the amygdala (Robson et al. 1998) (Seckl 1997). The SCO is involved with the central regulation of aldosterone secretion and sodium homoeostasis while the amygdala MR has been implicated in the control of salt appetite (Sakai et al. 1996). In contrast, 11β-HSD1 is much more widely distributed in the brain and appears to amplify glucocorticoid action in areas such as the hippocampus (Seckl 1997, Ajilore & Sapolsky 1999). The lack of 11β-HSD2 in MR-rich areas suggests that the majority of brain MR are likely to be occupied by glucocorticoid. Brain MR may play a key role in a number of homeostatic mechanisms including blood-pressure regulation, thirst and salt appetite, learning and memory, and hypothalamic/pituitary adrenal axis regulation, although it is often difficult to distinguish between glucocorticoid and mineralocorticoid effects on the MR.
Despite this, aldosterone-specific mediated effects have been shown by Gòmez-Sánchez (1996) who administered aldosterone in an i.c.v. manner to the rat brain at low doses that have no effects if given subcutaneously. Aldosterone, over a 4–5-week period, produced sustained dose-responsive increases in systemic blood pressure within 2 weeks, which appear to be independent of changes in sodium handling (Kageyama & Bravo 1988, Peysner et al. 1990). Concomitant i.c.v. infusion of corticosterone or the MR antagonist prorenone or RU-28318 attenuated i.c.v. aldosterone hypertension. Aldosterone seems to require a normal circulating background of corticosterone in order to exert these effects as bilateral adrenalectomy prevents the pressor effect of i.c.v. aldosterone, although it can be restored by corticosterone replacement (Gomez-Sanchez et al. 1990a, 1990b).
The CNS also appears to play a key role in the pressor effects caused by systemic mineralocorticoid excess. i.c.v. infusion of RU-28318 attenuated the hypertensive effect of subcutaneous aldosterone infusion or DOCA plus sodium administration. i.c.v. RU-28318 also normalized baroreflex activity (which is usually attenuated prior to blood-pressure elevation in this model) and reduced sympathetic tone (Gomez-Sanchez et al. 1990a, Janiak et al. 1990). In contrast, the prehypertensive stage of i.c.v. aldosterone induced hypertension is not associated with decreased sensitivity of the baroreceptor or altered sympathetic tone (Janiak et al. 1990). The prevention of hypertension by i.c.v. infusion of RU-28318 does not prevent the development of cardiac hypertrophy and fibrosis caused by systemic mineralocorticoid excess (Young et al. 1996). These results suggest that the CNS plays a significant role in the pressor effects of systemic aldosterone. These central effects appear to be distinct from its systemic effects on fluid/electrolyte balance and trophic effects on the vasculature and heart.
Non-genomic actions of aldosterone
The classical genomic model of aldosterone action in target epithelial cells has long been accepted. However, a number of recent studies have yielded data that are not explicable by this model and which suggest that aldosterone can also act by a rapid, non-genomic mechanism (Wehling 1997).
Non-genomic actions
The genomic actions of aldosterone described above occur through changes in gene expression and subsequent protein production that result in a lag time of 1–2 h before obvious changes in target cell activity. These effects can be inhibited by actinomycin and cycloheximide, which block transcription and translation respectively. In contrast, rapid effects that take place within 15 min have also been identified, predominantly in non-epithelial cells such as vascular smooth muscle cells, lymphocytes, endothelial cells, cardiac myocytes and kidney cells, but also in colonic epithelial cells. These effects are believed to be independent of gene transcription and translation as they are insensitive to actinomycin and cycloheximide; additionally, they were first identified in erythrocytes which lack nuclei (Falkenstein et al. 2000, Harvey et al. 2001, Losel et al. 2004).
The non-genomic receptor
Recent research into non-genomic effects has focused on identifying a specific non-genomic receptor, distinct from the MR, elucidating its cellular effects and mechanism of action, although there are suggestions that the effects may be mediated by the classical MR or a closely related protein (as has been shown for oestrogen and progesterone receptors). In rat arterial smooth muscle cells, rapid sodium efflux mediated by aldosterone is insensitive to actinomyocin, and therefore independent of gene transcription. It is sensitive to inhibition by spironolactone and RU-28318, implying the involvement of the classical MR. Similarly, eplerenone has been shown to inhibit the non-genomic effects of aldosterone on the NHE, [Ca2+]i levels and vasoconstriction in mesenteric resistance vessels (Michea et al. 2004). However, the great majority of studies have shown that MR antagonists are ineffective in blocking these effects. In addition, rapid actions of aldosterone on calcium efflux have been demonstrated in cell lines lacking the classical MR and effects on calcium and cAMP have been shown in skin fibroblasts from MR-knockout mice (Haseroth et al. 1999). Aldosterone also failed to elicit rapid effects in cells engineered to overexpress MR which under normal circumstances do not express MR and which do not exhibit rapid non-genomic responses (Falkenstein et al. 2000). These studies suggest that the rapid non-genomic effects of aldosterone are mediated by a distinct, novel receptor which, unlike the classical cytosolic/nuclear receptor, is associated with the cell membrane and has a high affinity for aldosterone but does not bind glucocorticoids. Although an approximately 50 kDa candidate membrane protein has been isolated from human lymphocytes, full structural characterization of this has yet to be achieved (Eisen et al. 1994).
Signal transduction and cellular effects
The non-genomic actions of aldosterone are varied and mediated by a multitude of second-messenger systems depending on the cell type involved. In vascular smooth muscle cells, aldosterone, like Ang II, increases IP3, DAG and subsequent [Ca2+]i (Christ et al. 1995). These second messengers then stimulate the translocation of PKC from the cytosol to the membrane. This protein appears to play a key role in the non-genomic mechanism. PKC inhibits the rapid activation of the Na+/H+ antiporter by aldosterone. PKC can also be activated independently of IP3 and DAG in colon cells, suggesting that it may actually be the non-genomic receptor. Aldosterone also increases cAMP levels in smooth muscle which activate protein kinase A. This seemingly opposing action of aldosterone on smooth muscle second-messenger systems, normally associated with both contraction and relaxation, may be a mechanism by which the cells are primed for rapid contraction/relaxation cycles. These contrasting second-messenger effects may also explain aldosterone’s action in colonic epithelia, where it rapidly activates ATP-sensitive K+ (KATP) channels but inhibits Ca2+-activated K+ channels. Final effectors of the non-genomic actions of aldosterone include K+ channels, NHE3 and other proteins involved in electrolyte and fluid balance and Ca2+ metabolism.
While many non-genomic effects have been demonstrated in vitro, the physiological impact of these is unclear. Non-genomic actions may be important to the responses mediated by local aldosterone production shown in the CNS and vascular system. A recent clinical study showed that administration of aldosterone significantly reduced forearm blood flow within 4 min, a time course far too rapid to be accounted for by classical genomic actions (Romagni et al. 2003). Other studies have shown a small but significant increase in systemic vascular resistance within 3 min of aldosterone administration (Wehling et al. 1998). Similar rapid aldosterone actions have been reported on heart rate, peripheral vascular resistence and baroreflex sensitivity (Weber & Purdy 1982, Yee & Struthers 1998, Schmidt et al. 1999).
Extra-adrenal aldosterone biosynthesis
It is now clear that many classic hormones are synthesized (or activated) in the tissues or cells where they act. The concept of local RASs is well established, with clear evidence to demonstrate generation of Ang II near its site of action (Samani 1994). Therefore, it has become necessary to ask whether corticosteroids can be synthesized in sites other than the adrenal cortex The production of neurosteroids (see below) is already well established and, with the advent of new, highly sensitive molecular techniques, it has become apparent that aldosterone may be produced in the CNS, as well as in the cardiovascular system (Davies & MacKenzie 2003). The evidence for extra-adrenal secretion of aldosterone, coupled with its known non-epithelial actions, has led to speculation that the hormone may act in such tissues in an autocrine or paracrine manner.
Cardiovascular system
In the last few years it has become apparent that many of the genes encoding enzymes and cofactors involved in aldosterone biosynthesis are also expressed in the cardiovascular system. Human aorta and pulmonary artery endothelial and smooth muscle cells were found to express the genes encoding P450 scc, 3β-HSD (types 1 and 2), 21-hydroxylase and aldosterone synthase, in addition to MR (Takeda et al. 1994). Transcripts for the cofactors StAR and adrenodoxin have also been detected, suggesting that all of the components necessary for local de novo biosynthesis from cholesterol are present, although an alternative possibility is that local aldosterone synthase acts on circulating precursors such as DOC. Regulation of gene expression has also been demonstrated and appears to be similar to that observed in the adrenal cortex, i.e. CYP11B2 expression in human umbilical vein endothelial cells is increased by Ang II and K+ (Takeda et al. 1996). It has been proposed that vascular aldosterone production may play a role in the aetiology of hypertension. For example, aortic CYP11B2 expression was reported to be increased in stroke-prone, spontaneously hypertensive (SHRSP) rats compared with Wistar-Kyoto (WKY) rats although this result has not been reproduced (Wu et al. 1998). In addition, CYP11B2 transcript levels are significantly higher in the mesenteric arteries of young SHRSP rats compared with WKY controls, although these differences do not persist into later life (Takeda 2004).
Conflicting data have been obtained in cardiac tissue. Delcayre and colleagues (Silvestre et al. 1998a) first described CYP11B2 expression in the four cardiac chambers of the adult Wistar rat; this was upregulated in response to Ang II or sodium restriction. The genes for P450 scc, P450c21 and type 2 3β-HSD and MR are also transcribed in all chambers of the normal human adult heart and fetal heart (Kayes-Wandover & White 2000). CYP11B2 transcription was detected in the fetal heart but not in normal adult cardiac chambers, implying that cardiac aldosterone expression may only be of significance under pathological conditions. Cardiac levels of CYP11B2 transcripts are reported to be raised during heart failure and a direct correlation has been identified between myocardial CYP11B2 mRNA expression and collagen volume has been shown in the failing human heart (Young et al. 2001, Satoh et al. 2002). After myocardial infarction, Wistar rats had increased cardiac levels of CYP11B2 transcripts and aldosterone in the non-infarcted areas of their left ventricles, although CYP11B1 mRNA and corticosterone levels actually fell in these areas (Silvestre et al. 1999).
Although these studies go some way towards proving the existence of local aldosterone biosynthesis, many of the results are inconsistent and the levels of gene expression are several orders of magnitude lower than those observed in the adrenal cortex. Indeed, using a highly sensitive and specific quantitative, real-time reverse transcriptase (RT)-PCR system we have been unable to detect CYP11B2 expression in any region of the rat heart under basal or pathological circumstances. Furthermore, whether these low levels of transcription result in translation and subsequent production of active enzyme is unclear. Studies at the protein level are scarce, perhaps reflecting the difficulties associated with such low levels of expression and the availability of specific antibodies.
However, there is evidence to suggest that local systems can produce significant concentrations of steroid. Aldosterone was detected in the perfusate of isolated mesenteric arteries from normotensive WKY rats, albeit at much lower levels than those found in the adrenal cortex or plasma (Takeda et al. 1995). This was significantly decreased by treatment with the ACE inhibitor, quinapril, and increased by perfusion with Ang II or potassium. Tissue aldosterone levels were significantly higher in the mesenteric arteries of young SHRSP rats than those of WKY animals (Takeda et al. 1997). Aldosterone levels are elevated in the failing human heart, exceeding circulating concentrations (Mizuno et al. 2001). Rat myocardial aldosterone concentration has been estimated at 16 nM, a level 17 times higher than in plasma, although others have found it not to differ significantly from plasma levels (Silvestre et al. 1998b). Such a high local concentration has been attributed to slower aldosterone degradation in the heart, to intracellular sequestration or to local delivery of aldosterone to extracellular spaces rather than into the bloodstream (Silvestre et al. 1998a). However it is achieved, such levels of aldosterone may compensate for the low levels or absence of cardiac 11β-HSD2, and facilitate the occupation of MR by mineralocorticoid rather than glucocorticoid. As a cautionary note, it should be added that a recent study found cardiac aldosterone to be present at levels 14–28 times lower than the 16 nM estimate (Gomez-Sanchez et al. 2004). Similarly, our own studies of isolated rat cardiomyocytes have failed to detect any aldosterone synthase activity.
Thus, the evidence for local production of aldosterone within cardiac tissue remains contradictory. While the non-epithelial actions of aldosterone, particularly in the heart, are clear, there is no direct evidence that these are achieved through local rather than adrenal production. Indeed, Ang II-/NaCl-induced cardiac fibrosis is reversed by adrenalectomy, suggesting that the effects in this model are mediated by adrenal aldosterone (Rocha et al. 2002). Furthermore, it may be of relevance that the recently described aldosterone synthase-knockout mouse has no reported defect in cardiac structure or function despite possessing no detectable aldosterone in its circulation (G Lee 2004, personal communication).
CNS
Steroid biosynthesis in the CNS is a well-documented phenomenon; the term neurosteroids was devised to describe steroids synthesized from cholesterol and other early precursors, e.g pregnenolone and dehydroepiandrosterone (DHEA; Baulieu & Robel 1990). This finding resulted predominantly from the immunohistochemical detection of P450SCC in glial cells which can convert cholesterol to pregnenolone in vitro (Jung-Testas et al. 1989). However, it is now clear that that the CNS may also be capable of producing the corticosteroids, including aldosterone. CYP11A1 transcripts (or P450 scc itself) have been found in neurons such as the Purkinje cells of the rat cerebellum (Furukawa et al. 1998, Ukena et al. 1998) and fetal rat primary hippocampal cells (Sanne & Krueger 1995). Transcripts for StAR protein have also been detected in the adult rat CNS by RNase protection assay, RT-PCR and in situ hybridization (Furukawa et al. 1998). They are abundant in the cerebral cortex, the pyramidal cell layers of the hippocampus and dentate gyrus, and the olfactory bulb. The cerebellum has the highest levels of all. Virtually all rat tissues that have been examined, including the brain, contain adrenodoxin mRNA (Mellon et al. 1991, Stromstedt & Waterman 1995, MacKenzie et al. 2000a). 3β-HSD has been localized to the cerebral cortex, hippocampus, cerebellum, thalamus, hypothalamus and olfactory bulb. The highest levels of 3β-HSD expression within the hippocampus are found within the CA1–2 fields and the dentate gyrus whereas, in the cerebellum, expression is strongest in the Purkinje cells, granule cells and stellate/basket cells. There have been fewer investigations of 21-hydroxylase but mRNA has been detected in the brains of mice and humans (Stromstedt & Waterman 1995, Beyenburg et al. 2001). Thus all of the key genes involved in corticosteroid biosynthesis are expressed at fairly high levels in specific CNS regions.
Aldosterone synthase gene expression has been detected in whole rat brain and in cortex, cerebellum, brain stem, hippocampus, hypothalamus and amygdala homogenates (Albiston et al. 1994, Gomez-Sanchez et al. 1996). Significantly, we have shown that CYP11B2 expression in the hippocampus and cerebellum is regulated by the peripheral RAS; sodium depletion increases CYP11B2 expression in these areas, in parallel with similar changes in the adrenal ZG (Ye et al. 2003). In addition, we have shown that CYP11B2 is also expressed in the human brain, (S M MacKenzie and E Davies, unpublished observations). Once again, levels of gene expression in the CNS are much lower (100–1000-fold) than in the adrenal cortex. However, we have verified protein synthesis by immuno-histochemistry, and demonstrated aldosterone synthase production in the hippocampus and cerebellum (particularly the Purkinje cells; MacKenzie et al. 2000a). We also demonstrated positive immunostaining for aldosterone synthase in primary cultures of fetal rat hippocampal neurons (MacKenzie et al. 2002b). Interestingly, aldosterone synthase localizes to precisely the same areas of the brain as 11β-hydroxylase, CYP11A1, 3β-HSD and StAR, shown by in situ hybridization (Furukawa et al. 1998), and correlates well with areas of high MR expression (Agarwal et al. 1993). This is consistent with an autocrine or paracrine model of corticosteroid action in the brain. Further evidence comes from studies showing that brain homonogenates can convert DOC to corticosterone, 18-hydroxy-deoxycorticosterone and aldosterone (Gomez-Sanchez et al. 1996). In addition, rat hippocampal neurons can convert the corticosteroid precursor DOC to aldosterone and corticosterone in vitro, although de novo synthesis from cholesterol has yet to be demonstrated (MacKenzie et al. 2000b).
Thus there is strong evidence to support the production of aldosterone in the CNS and progress has been made in identifying regulatory factors that influence local production. However, it is much more difficult to establish whether locally produced aldosterone exerts any significant physiological or pathophysiological effects. It is noteworthy that i.c.v. infusion of 19-ethynyl-DOC, an irreversible inhibitor of aldosterone synthase, in the Dahl salt-sensitive hypertensive rat at systemically insignificant doses reduces salt-induced hypertension (Gomez-Sanchez et al. 1996). This suggests that aldosterone production within the brain regulates blood pressure in this genetic model of hypertension (Gomez-Sanchez et al. 1990b). The SCO – one of the few adult brain regions to demonstrate MR selectivity due to its expression of 11β-HSD2 – has been implicated in this central hypertensive effect.
Aldosterone: a key cardiovascular hormone
The above descriptions of aldosterone’s influence over cardiovascular function (through a range of cardiac, renal, vascular and CNS actions) identify its potential to play an important role in pathophysiological circumstances. The potential importance of this has been increasingly appreciated over the last few years.
Aldosterone deficiency
The consequences of deficient aldosterone synthesis are illustrated in the rare syndromes associated with loss of function of the CYP11B2 gene. A detailed review of this is beyond the scope of this article but is summarized fully in White (2004). In brief, inactivating mutations of aldosterone synthase lead to failure of aldosterone synthesis in neonatal life (CMO II deficiency). Affected infants present with hyperkalaemia, salt wasting and failure to thrive; treatment with increased sodium intake and mineralocorticoid supplementation provides effective treatment. In older subjects the phenotype becomes less severe, and there appear to be no other major cardiovascular consequences. This human disorder has recently been reproduced in the aldosterone synthase-knockout mouse model; initial descriptions of this indicate that animals have loss of aldosterone synthesis. In addition to the expected biochemical and electrolyte phenotype, animals have lower blood pressure than wild-type littermates (G Lee 2004, personal communication).
A similar amelioration of phenotype with aging is observed in the autosomal dominant form of pseudohypoaldosteronism (PHA) type 1, where aldosterone activity is abolished due to mutations within MR (Sartorato et al. 2004). As with CMO II deficiency, affected neonates present with salt wasting and hyperkalaemia, but in later life have little overt electrolyte disturbance. These two rare conditions illustrate the key importance of aldosterone action to electrolyte homoeostasis in early life; in older subjects, additional mechanisms appear to reduce the absolute dependency on aldosterone action for Na+ and K+ regulation.
The other common circumstance in which aldosterone is deficient is Addison’s disease. Here, aldosterone deficiency is generally seen in the context of failure of glucocorticoid synthesis; adult patients display the classical phenotype of volume deficiency, hypotension, hyponatraemia and hyperkalaemia, reflecting mineralocorticoid deficiency. However, it is clear that glucocorticoid deficiency also contributes to this presentation; adult patients who have genetically determined defects in the MR (autosomal dominant PHA type 1) but have normal glucocorticoid production and action have a much less severe clinical phenotype (Zennaro & Lombes 2004).
Aldosterone excess
Overproduction of aldosterone is a more common clinical circumstance than deficiency. It is associated with a range of severe cardiovascular consequences that follow from the aldosterone actions described earlier. The classic features of aldosterone overproduction are seen in the syndromes of primary aldosteronism (PA), first described by Jerome Conn in 1954 (Conn & Louis 1956) Affected subjects present with high blood pressure that is generally resistant to conventional antihypertensive therapies. There is expansion of the exchangeable body Na+ content and an increase in plasma volume. Total body K+ content and plasma K+ concentration are both reduced. However, more recent understanding of the biology surrounding aldosterone’s actions has led to an appreciation that aldosterone excess has significant deleterious effects on several cardiovascular tissues. Thus, patients with PA show evidence of cardiac hypertrophy that is excessive in relation to the prevailing level of blood pressure but consistent with aldosterone-mediated remodelling of cardiac tissue. Additionally, patients with the form of aldosterone excess termed glucocorticoid-remediable aldosteronism (GRA) are reported to have an excess risk of intracranial aneurysms and subarachnoid haemorrhage in early life, an association that suggests aldosterone has chronic adverse effects on cerebrovascular function (Litchfield et al. 1998). Other evidence of deleterious tissue consequences comes from animal models of mineralocorticoid excess. Here, studies show that rodents given high doses of DOC or aldosterone develop a very marked perivascular inflammation in cardiac, renal and cerebral vessels (Rocha & Funder 2002). These changes are associated with increased collagen synthesis and scarring. It is of interest that these aldosterone-mediated effects are dependent on a concomitant high sodium intake. The biological reason for this is uncertain, and it is not known whether there is a similar relationship in humans between sodium status and mineralocorticoid-related damage (Funder 2004).
PA is defined as excessive production of aldosterone independent of its normal trophin (e.g. Ang II). The previously mentioned GRA is a rare autosomal dominant form of PA accounted for by a chimaeric CYP11B1/ CYP11B2 gene that encodes aldosterone synthase but is regulated in its expression by the CYP11B1 5′UTR, which is responsive to ACTH (Lifton et al. 1992). In this circumstance, aldosterone synthase is expressed in the zona fasciculata. Affected subjects make excessive aldosterone from early life; the production of aldosterone can be suppressed by inhibition of ACTH secretion with exogenous glucocorticoid. This rare disorder has a notably variable phenotype; some subjects possess severe hypertension from an early age, whereas others have a much milder form. This variability can be encountered within kindreds who share the same causal mutation, and may reflect epistatic interaction with other loci that affect cardiovascular function.
The common clinical forms of PA are accounted for by adrenal nodular disease. In a minority of subjects, a unilateral adrenal adenoma is present, while the majority of patients with PA have bilateral adrenal hyperplasia (idiopathic hyperaldosteronism). Earlier studies indicated that PA, of whatever subtype, was a relatively rare disorder. However, studies in recent years from a wide geographic representation have demonstrated that around 10% of unselected subjects with hypertension have evidence of aldosterone excess (Mulatero et al. 2004). Many of these studies have relied on measurement of the ratio of aldosterone to renin as a screening test for the presence of PA. This has been subject to criticism, on the basis that the test has not been standardized or evaluated formally (Montori & Young 2002). However, the evidence that PA is a relatively common disorder is supported by studies that have used a range of other detection and confirmation methods to identify PA. For example, in a recent large study of a German population, Abdelhamid et al.(2003) established that up to 20% of patients had aldosterone excess by using a very sensitive measurement of urinary tetrahydroaldosterone excretion, combined with a range of dynamic endocrine tests and imaging. Thus the weight of evidence is such that there can be no doubt that a significant percentage of patients with hypertension do have excessive aldosterone secretion, inappropriate for the prevailing levels of renin and Ang II. In such patients, who probably represent at least 10% of the hypertensive population, it is reasonable to speculate that aldosterone is contributing to the high blood pressure and the associated tissue damage. There remains some doubt as to the absolute proportion of such subjects who harbour a unilateral adrenal adenoma, the removal of which will cure or greatly ameliorate the hypertension. Young (2003), presenting data from the Mayo Clinic, estimated that such patients were a minority of the total PA population. Other authors have suggested that assiduous search for lateralizing adrenal adenomas is justified on the basis that these are the most common form of PA. A full review of this controversy is not appropriate here, but the reader is referred to several recent reviews and debates on the subject (Lim et al. 2002, Padfield 2002, Stowasser & Gordon 2004).
The underlying reason for the development of PA is unresolved. Many years ago it was proposed that there was no substantial difference between patients with PA due to bilateral adrenal hyperplasia and subjects with low-renin essential hypertension – both groups had expansion of exchangeable body sodium content, suppression of renin and aldosterone production that was inappropriately high for the level of renin (Padfield et al. 1981). In both groups aldosterone was still responsive to the stimulatory effect of Ang II. These similarities of phenotype suggest that, in many patients, PA may be a subtype of hypertension in which aldosterone production has become dysregulated, so that it is excessively sensitive to its usual trophin. Whether it is truly autonomous in this circumstance is probably a matter of semantics; the true importance lies in the recognition that aldosterone is excessive and is responsible for cardiovascular dysfunction that can be treated by selective MR antagonism. It is also unclear whether patients who have unequivocal bilateral adrenal hyper-plasia ever progress to a stage where a single unilateral nodule becomes dominant and responsible for the aldosterone excess (the analogy with thyroid nodular disease is pertinent here). However, histological studies that demonstrate the remainder of the adrenal cortex in patients with an apparent aldosterone producing adenoma is abnormal (hyperplastic), suggest that the solitary lesion arises on a background of a more diffuse disorder. If correct, it may be that there is a spectrum of disorders leading to the phenotype of PA that can, in some circumstances, show transition of one to the other.
We have proposed previously that PA might be a disorder that develops slowly over many years, reflecting an interaction of genetic susceptibility and environmental factors (Connell et al. 2003). We initially reported that a polymorphic variation (−344 C/T) in the 5′UTR of the CYP11B2 gene was associated with increased secretion of aldosterone, a finding that has been supported by other groups (Paillard et al. 1999). Since then we have found that the same polymorphism (T allele) is associated with a higher prevalence of hypertension (Davies et al. 1999). This finding has been corroborated by some other studies, but by no means all of them (Brand et al. 1998, Paillard et al. 1999, Tsujita et al. 2001). We also found that within a hypertensive population subjects with the same polymorphic variation (T allele) had a higher frequency of PA, and that that this allele was also over-represented in a population with adenomatous PA (Inglis et al. 2001). Thus there is evidence that a variation in the aldosterone synthase gene is associated with development of PA. However, the intermediate phenotype and mechanism that accounts for this is less clearly defined. In a series of studies we have shown that the T allele of the −344 C/T polymorphism is most strongly associated with increased secretion of the 11-deoxysteroids, 11-deoxycortisol and DOC (Davies et al. 2001). Again, this is consistent with data from other independent groups (Hautanena et al. 1998). Most recently, we have shown that this polymorphism accounts for a significant proportion of the variability of 11-deoxycortisol and DOC production in a normal population (Kennon et al. 2004). This finding initially appears paradoxical: the phenotype of increased production of these 11-deoxysteroids is consistent with reduced efficiency of the enzyme 11β-hydroxylase, encoded by the CYP11B1 gene. However, we have now identified that the −344 C/T polymorphism of CYP11B2 is in close linkage dysequilibrium with several polymorphisms in CYP11B1 which may account for the altered 11-hydroxylase efficiency. Whether the polymorphisms in CYP11B1 also account for the association of the locus with hypertension (particularly mineralocorticoid hypertension) remains to be resolved. However, it is pertinent that variation in the same gene (encoding 11β-hydroxylase) accounts for the biochemical and cardiovascular phenotype in the Dahl salt-sensitive hypertensive rat (Cicila et al. 1993, Garrett & Rapp 2003).
These data raise questions about the long-term development of PA and we have proposed a possible mechanism in this regard (Connell et al. 2003, Freel & Connell 2004). In short, a genetically determined and lifelong reduction in the efficiency of 11-hydroxylation will result in a small increase in DOC and 11-deoxycortisol, which are unlikely to have any significant biological effect. However, the resulting decrease in cortisol production will be compensated for by a small increase in ACTH drive to the adrenal cortex to maintain normal production. As a consequence, over a period of many years, the adrenal will be exposed to a hyperplastic influence of this peptide. We have speculated that, in contrast to previous studies which showed a decrease in aldosterone production following chronic stimulation with supraphysiological concentrations of ACTH (Oelkers 1985), this subtle increase in ACTH over time leads to increased sensitivity of aldosterone to a range of trophins, including Ang II and K+. In subjects with high Na+ intake (and possibly in the context of other permissive genetic variations) this will progress to an eventual phenotype of PA with bilateral adrenal hyperplasia. The evidence cited above concerning the influence of ACTH on normal regulation of aldosterone synthesis from the POMC-knockout mouse lends some support to this hypothesis (Coll et al. 2004). In addition, adrenal gland hyperplasia and defective 11β-hydroxylation have been described in patients with hypertension (Symington 1969, de Simone et al. 1985).
These speculations on the origins and definition of PA do not detract from the importance of aldosterone as a key hormone in patients with hypertension. Recognition of this acknowledges the possibility of wider use of mineralocorticoid receptor antagonists in the treatment of hypertension. The efficacy of spironolactone in the treatment of patients with PA or in patients with a high ratio of aldosterone to renin is well established (Lim et al. 1999). However, the use of this agent is associated with a high level of unwanted effects (loss of libido; gynaecomastia) in male patients due to its affinity for other steroid receptors. The development of Eplerenone, a more-selective aldosterone receptor antagonist that lacks these unwanted effects, offers the possibility of more extensive use of this class of drugs. At present, experience with Eplerenone in hypertension shows it to be effective in lowering blood pressure in a range of patient populations. Importantly, this drug therapy results in regression of left-ventricular hypertrophy, an effect that is additive when prescribed in addition to an ACE inhibitor (Liew & Krum 2003, Pitt et al. 2003a).
Heart-failure syndromes
The role of aldosterone excess in the development and progression of heart failure has been recognized for many years. In this circumstance, aldosterone excess is secondary to activation of the neuro-humoral axis, and contributes to excess sodium retention and vascular dysfunction that are characteristic features. However, the impact of aldosterone on vascular function is difficult to dissect from the deleterious effects of other hormones such as Ang II. There are data which show that aldosterone reduces nitric oxide availability and increases production of molecules that promote atherogenesis, such as plasminogen activator inhibitor (PAI1; Brown et al. 2002). In addition, there is some evidence that aldosterone increases the expression of vascular ACE, so that the generation of Ang II is enhanced (Macdonald et al. 2004). Thus there may be positive feedback that leads to aldosterone increasing the production of, and interacting with, Ang II to exacerbate vascular dysfunction; it is reasonable to assume that aldosterone and other hormones such as Ang II act in synergy to cause the cardiac, renal and vascular dysfunctions that occur in heart failure.
The evidence that aldosterone contributes to the development and progression of heart failure implies that antagonism of its action would be beneficial. There is a large body of data that illustrates the benefit of ACE inhibition and Ang II receptor antagonism in heart failure (Pitt et al. 2000). Such therapies might be expected to oppose the adverse effects of aldosterone by limiting its production. However, there is good evidence that aldosterone ‘escapes’ from ACE inhibition or Ang II receptor blockade in chronically treated heart-failure patients, so that circulating levels are not significantly reduced. In part, this illustrates the lack of dependence on a single trophin to maintain aldosterone secretion – in circumstances where Ang II receptor activation is inhibited, changes in extra-cellular potassium ion concentration will still have a major role in regulating aldosterone secretion. The example of the angiotensinogen-knockout mouse is consistent with this notion; the model has no detectable Ang II generation, but can regulate aldosterone normally to maintain potassium K+ homoeostasis, confirming that angiotensin availability is not essential in production of aldosterone (Okubo et al. 1998).
The persistence of aldosterone secretion in treated heart-failure patients led to the hypothesis that aldosterone blockade might provide additional therapeutic benefit over and above existing therapies. The RALES study provided convincing evidence that this was the case (Pitt et al. 1999). In this large study of 3400 patients with severe heart failure, patients randomized to spironolactone in addition to existing conventional therapy showed a 30% reduction in mortality. Interestingly, much of this benefit was seen fairly soon after randomization, and was due to a reduction in sudden cardiac death, suggesting that aldosterone blockade reduced the incidence of cardiac rhythym disturbance. Following this study, the EPHESUS trial examined the effect of treatment with Eplerenone in patients following acute myocardial infarction (Pitt et al. 2003b). Patients randomized to active therapy in this study showed a 15% reduction in mortality. Again, the effects were noted quite early after randomization, before cardiac remodelling might have been expected to have occurred. This may suggest that aldosterone receptor blockade produces a benefit by altering the risk of cardiac electrical disturbance. There are a number of ways in which this might occur, including altered electrolyte homoeostasis, change in central sympathetic outflow, altered baroreflex activity and improved vascular endothelial function. However, regardless of the exact mechanism, there is now very persuasive evidence that selective aldosterone receptor antagonism (SARA) offers significant advantage in patients with secondary hyperaldosteronism or following myocardial infarction. This, in turn, is consistent with the suggestion that aldosterone has major deleterious effects on cardiovascular function and mortality in these circumstances.
Future perspectives
In the last few years much has been learned about aldosterone synthesis and action. The role of novel adipose-derived factors that contribute to aldosterone regulation offers a novel explanation for the overproduction of aldosterone that occurs in hypertension. However, the significance of this mechanism in human physiological regulation is a topic that requires future study. Research into the action of aldosterone has challenged the original belief that aldosterone acts solely on specific receptors in epithelial tissues and modulates electrolyte and water balance via a genomic mechanism. Indeed, extra-adrenal biosynthesis, alternative non-epithelial target tissues and rapid non-genomic mechanisms of action have now been revealed. It is likely that there is synergy between the classic effects of aldosterone and these novel mechanisms; future studies to dissect the precise pathways that account for the integrated action of aldosterone are awaited. Despite this wide range of target tissues and mechanism of actions, all of its effects are targeted towards regulation of the cardiovascular system and its role as a key cardiovascular hormone is now recognized. However, there remains considerable uncertainty over the way in which aldosterone acts to determine cardiac hypertrophy. While the presence of specific mineralocorticoid receptors in cardiac tissue is well established, the way in which these can remain selective for aldosterone in the presence of excess glucocorticoid remains to be identified. Potential mechanisms have been described above. Additionally, studies to determine the exact role of local synthesis of aldosterone in determining the mineralocorticoid regulation function of the CNS and vascular tissue are indicated. In this regard the aldosterone synthase-knockout mouse is likely to provide valuable information.
In clinical studies the importance of aldosterone in hypertension remains the subject of debate. While evidence suggests that this may be a factor in around 10% of patients with high blood pressure, the pathophysiological mechanisms that underlie this are unclear and are likely to be the subject of future research. Additionally, the value of aldosterone receptor blockade in the management of hypertension is the subject of current research studies that are likely to report in the next few years. Finally, the importance of aldosterone as a hormone that is key in the development and progression of heart failure has been illustrated by the findings from the RALES and EPHESUS studies, but the way in which aldosterone anatagonism is best employed remains to be clarified, as does the impact of new agents such as Eplerenone on cardiac fibrosis in humans. While current therapies focus on MR antagonism, an alternative approach is to inhibit aldosterone synthesis by selective inhibitors of aldosterone synthase. Such agents are currently in development, but their clinical utility is not clear.
In summary, research into aldosterone synthesis, action and clinical importance has reached an exciting phase. Recent advances have raised a number of questions about the way in which the hormone acts to cause cardiovascular damage in hypertension and heart failure and their resolution is likely to lead to new and better treatments for these syndromes. The next 50 years promise to be of considerable interest.
Acknowledgments
The authors would like to thank Dr Scott MacKenzie for critical review of the manuscript and preparation of the figures. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
- Received 30 September 2004
- Accepted 2 December 2004
- © 2005 Society for Endocrinology
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵










