- Made available online as an Accepted Preprint 10 March 2010
- Accepted Preprint first posted online on 10 March 2010
Role of the high mobility group A proteins in the regulation of pituitary cell cycle
- 1Istituto di Endocrinologia ed Oncologia Sperimentale (IEOS) del CNR, 80131 Naples, Italy
2Dipartimento di Biologia e Patologia Cellulare e Molecolare, Università degli Studi di Napoli ‘Federico II’, 80131 Naples, Italy
- (Correspondence should be addressed to M Fedele at Dipartimento di Biologia e Patologia Cellulare e Molecolare, Istituto di Endocrinologia ed Oncologia Sperimentale (IEOS) del CNR, Università degli Studi di Napoli ‘Federico II’; Email: mfedele{at}unina.it)
Abstract
Pituitary cells are particularly sensitive to alterations of the cell cycle machinery. In fact, mutations affecting expression of proteins critical for cell cycle progression, including retinoblastoma protein, cyclins D1 and D3, p16INK4A, and p27kip1, are frequent in human pituitary adenomas. Similarly, both targeted disruption and overexpression of either cell cycle inhibitors or activators, respectively, lead to the development of pituitary adenomas in mice. Recent evidence has added the high mobility group A (HMGA) proteins as a new class of cell cycle regulators that play significant roles in the pathways that lead to pituitary tumor evolution in both humans and experimental animal models. Here, we first review the role of the cell cycle in pituitary tumorigenesis, as witnessed by human pathology and transgenic mice; and then, we focus on HMGA proteins and their cell cycle-related role in pituitary tumorigenesis.
Introduction
The pituitary gland is a critical endocrine organ on which growth, metabolism, reproduction, and homeostasis depend. In mammals, it consists of three lobes: the anterior and the intermediate lobes form the adenohypophysis, while the posterior lobe represents the neurohypophysis. The adenohypophysis secretes seven different hormones: GH, TSH, prolactin (PRL), FSH, LH, ACTH, and melanocyte-stimulating hormone, whereas the neurohypophysis releases two hypothalamic neuropeptides, vasopressin and oxytocin. In the adenohypophysis, hormones are produced by different cell types, whose absolute and relative number may vary during adult life depending on the endocrine demands (Sasaki 1988, Levy 2002). The regulation of the proliferative ability of pituitary cells in adulthood is not well established, although the presence of a side population of stem/progenitor cells, which may contribute to cell renewal in the adult pituitary, has been described (Chen et al. 2005, 2009). Recent experimental data suggest that cell cycle regulators may have significant implications in the biology of putative progenitor cells and pituitary homeostasis (Janzen et al. 2006, Macias et al. 2008). Indeed, cell cycle plays a key role in the development of pituitary adenomas as assessed in humans and mice.
The cell cycle
The cell cycle is the process by which cells divide into daughter cells. It comprises a defined sequence of events, which include: synthesis of DNA (S phase), mitosis (M phase), and two phases of growing and preparation for following steps (G1 and G2 phases; Fig. 1). A reversible quiescence phase (G0), in which cells ‘take a rest’ in the absence of mitogenic signals, is also considered part of the cell cycle (Norbury & Nurse 1992). At particular stages of the cell cycle, cells encounter a restriction point and four checkpoints that enable the correct progress and completion of each cycle. The restriction point (R) is a specific molecular event, which is set between early and late G1 phases, after which cells can proliferate independently from mitogenic stimuli (Pardee 1974), whereas checkpoints are surveillance mechanisms and quality controls of the genome to maintain genomic integrity, which are set between G1 and S, intra-S, between G2 and M and intra-M (Hartwell & Weinert 1989).
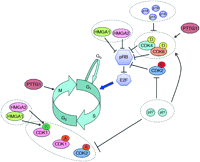
Schematic representation of the main cell cycle regulating pathways affected in pituitary tumors. The blue arrow indicates the restriction point. A, B, D, and E define the different cyclins. Stimulatory or inhibitory effects are indicated by an arrow or a bar, respectively, at the end of a line. Dotted ovals indicate group proteins and protein complexes that share similar functions and regulations.
Both activating and inhibitory signals, represented by cyclins coupled to cyclin-dependent kinases (CDKs) and CDK inhibitors (CKI), respectively, finely regulate the progress of the cell through the different phases of the cell cycle (Morgan 1997, Vidal & Koff 2000).
Among the activators, D-type cyclins, coupled to CDK4 and CDK6, promote the progression through G1; E-type cyclins, coupled to CDK2, are involved in the G1/S transition; A-type cyclins, coupled to CDK2 and CDK1, participate in the S/G2 transition and progression through G2 respectively; B-type cyclins, coupled to CDK1, are involved in the G2/M transition; cyclins A and B also complex with CDK1/CDC2/p34 to form the mitosis-promoting factor whose activity peaks at G2/M transition, being required for the cell to enter M phase (Pines & Hunter 1989).
The transcription factor family E2F plays a critical role in G0/G1, R, and G1/S transitions, by allowing transcription of D- and E-type cyclins. Consistently, the gatekeeper retinoblastoma protein (pRB) negatively regulates G1 entering, progression, and transition to S by blocking the activity of factors responsible for cell cycle progression, which include the E2Fs. CDK4, CDK6, and CDK2, complexed to D- and E-type cyclins, mediate phosphorylation of pRB, thus provoking the release and activation of E2F factors from pRB (Fig. 1). The pRB/E2Fs complex can also be regulated by acetylation/deacetylation, mediated by histone acetylases and deacetylases (Magnaghi-Jaulin et al. 1998, Martínez-Bálbas et al. 2000).
The action of the cyclin-CDK complexes is counteracted by specific CKIs either by blocking the action of CDKs or by impairing substrate/ATP access. The CKIs belong to two families: the INK4 and the Cip/Kip families. The INK4 family consists of four members (p16INK4a, p15INK4b, p18INK4c, and p19INK4d), which inhibit the G1/S transition by blocking CDK4 and CDK6. Conversely, the Cip/Kip family, which includes p21Cip1, p27Kip1, and p57Kip2, has different roles depending on the cyclin-CDK complexes, which they bind to. Association with cyclin-CDK2 and cyclin-CDK1 blocks their activity, whereas binding to cyclin-CDK4 and cyclin-CDK6 can upregulate their activity (Cheng et al. 1999, Sherr & Roberts 1999; Fig. 1).
Negative regulation of the cell cycle is not restricted to CKIs. One critical target of negative regulation that acts in M phase is a protein named securin or PTTG1 (Pei & Melmed 1997). During metaphase, replicated paired sister chromatids are held together by the cohesin complex, which is degraded by the proteolytic protein separase, during the metaphase to anaphase transition, to allow the separation of sister chromatids to proceed to diploid daughter cells (Uhlmann et al. 1999). The activity of separase is inhibited by the interaction with securin, whose proteosomal degradation by the ubiquitin ligase APC is crucial for the completion of the anaphase process (Zou et al. 1999). In addition to this securin function, recent reports indicate a role for PTTG1 in modulation of the G1/S phase transition by interacting with Sp1 and regulating the transcriptional activity on the cyclin D3 promoter (Tong et al. 2007). Interestingly, securin/PTTG1 is regulated by CDK1-mediated phosphorylation (Holt et al. 2008) suggesting a link between the control of the cell cycle by CDKs and securin function.
Cell cycle dysregulation in human pituitary tumorigenesis
Cell cycle dysregulation is the main pathogenetic event in the development of pituitary tumors. In fact, it has been estimated that more than 80% of human pituitary tumors display alterations at least in one of the regulators of the G1/S transition of the cell cycle (Malumbres & Barbacid 2001; Table 1). These alterations are frequently represented by epigenetic events that target several cell cycle regulators, leading to overexpression of cyclins (mainly D1, D3, E, B1, and B2), as well as to downregulation of CKIs (mainly p16INK4A, p15INK4B, p27Kip1, and p21Cip1) and pRB expression (Farrell & Clayton 2003). Cyclins D1 and D3, as well as cyclin E, are frequently overexpressed in all subtypes of human pituitary adenomas, with prevalence in nonfunctional pituitary adenomas (NFPA) for the D-type (Jordan et al. 2000, Turner et al. 2000, Saeger et al. 2001, Simpson et al. 2001) and in corticotroph tumors for the E-type cyclins (Jordan et al. 2000). A cyclin D1 gene allelic imbalance has also been described in about 25% of analyzed adenomas (Hibberts et al. 1999). B-type cyclins have recently been described as overexpressed in many human pituitary adenomas, with prevalence in prolactinomas (Wierinckx et al. 2007, De Martino et al. 2009b). Silencing of the gene encoding p16INK4A by methylation is frequent in pituitary, mainly NFPA (Woloschak et al. 1997, Simpson et al. 1999a, Morris et al. 2005, Ogino et al. 2005, Yoshino et al. 2007). Downregulation of p27kip1 protein expression, likely due to a JAB1-mediated increased proteolysis (Korbonits et al. 2002), is frequent in pituitary carcinomas and ACTH-secreting adenomas (Bamberger et al. 1999, Lidhar et al. 1999). Conversely, downregulation of p21Cip1 in pituitary adenomas may be due to epigenetic modifications (Yoshino et al. 2007, Zhu et al. 2008). Similarly, frequent loss of pRB expression observed in 27% of somatotrophinomas (Simpson et al. 1999b) was mainly due to methylation and/or microdeletion of the RB1 gene (Simpson et al. 2000).
Major cell cycle-related proteins altered in human pituitary tumors
In addition to these classical cell cycle-related proteins, the overexpression of PTTG1 has been observed in more than 90% of all types of pituitary tumors (Zhang et al. 1999). Finally, as described in more detail in a subsequent section of this review, overexpression of high mobility group A (HMGA) proteins has been shown to play a critical role in development of pituitary adenomas, mainly prolactinomas and GH/PRL-secreting adenomas, by mechanisms involving dysregulation of cell cycle-related proteins (Fedele et al. 2002, 2006, Finelli et al. 2002, De Martino et al. 2009b).
Cell cycle-related mouse models of pituitary adenomas
In keeping with the critical role of cell cycle dysregulation in pituitary tumorigenesis, targeted disruption or overexpression of cell cycle regulators frequently causes onset of pituitary adenomas (Table 2). The first mouse model of cell cycle-related pituitary tumorigenesis came from the phenotype of knockout mice for pRb (Jacks et al. 1992). The heterozygous animals developed pituitary tumors of the intermediate lobe (IL) arising from cells in which the wild-type Rb allele was absent. Loss of either E2F1 or E2F4 reduced pituitary tumorigenesis in these mice, suggesting that loss of pRB induces the onset of pituitary tumors by activating E2F factors (Yamasaki et al. 1998, Lee et al. 2002). Consistently, deregulated E2F activity in E2f3-transgenic mice induces hyperplasia of the pituitary gland (Lazzerini Denchi et al. 2005). Subsequently, three different groups reported the characterization of another mouse model of pituitary tumorigenesis: the knockout mice for p27kip1 (Fero et al. 1996, Kiyokawa et al. 1996, Nakayama et al. 1996). As in the pRb mutants, p27kip1-null mice develop pituitary adenomas involving IL melanotrophs, even though differences in terms of gene expression and tumor phenotypes have been observed between the two models (Chien et al. 2007). Therefore, despite the fact that deletions of p27Kip1 or Rb cause IL tumors in murine models, they may have different mechanisms of tumor suppression. Indeed, as already described above, in addition to cyclin D complexes, which act upstream of pRB, p27Kip1 inhibits both Cdk1 and Cdk2, both of which are capable of phosphorylating substrates other than pRB (Fig. 1). Crossing of pRb+/− and p27−/− mice lead to double mutants having a significantly reduced tumor latency period compared with single mutants (Park et al. 1999). Intriguingly, a recent new mouse model of pituitary tumorigenesis, the knock-in mice depleted of the Cdk inhibitory function of p27, develops pituitary adenomas larger and more vascular, causing more damage to surrounding tissue than those developing in p27-null mice (Besson et al. 2007). Interestingly, a recent study highlighted the importance of the mouse strain on pituitary tumor evolution, since it showed that 129Sv animals are inherently predisposed to IL pituitary tumors and that Rb+/− mice in a C57BL/6 background develop high-penetrance anterior pituitary (AP) tumors (Leung et al. 2004).
Mouse models of pituitary tumorigenesis caused by cell cycle dysregulation
Another Cdk inhibitor, p18ink4c, has been shown to be crucial in the homeostasis of the pituitary cell cycle. In fact, p18−/− mice develop pituitary adenomas in both intermediate and anterior lobes (Franklin et al. 1998). Interestingly, crossing of p18-null mice with mice deficient for other Cdk inhibitors, such as p16, p27, or p21, led to double mutant mice with increased incidence and decreased latency in the development of pituitary adenomas as compared with single mutant mice (Franklin et al. 1998, 2000, Ramsey et al. 2007). Therefore, the existence of two major pathways for G1/S phase dysregulation in pituitary tumors has been suggested: one involving p18/CDK4/pRB, and the other one involving p27 and p21 (Quereda & Malumbres 2009). Consistently, knock-in mice, which express a Cdk4 mutant originally found in human melanoma (Wölfel et al. 1995) and which are insensitive to INK4 inhibitors (Cdk4 R24C), develop several tumors, including pituitary adenomas (Sotillo et al. 2001, Rane et al. 2002). Crossing of Cdk4R24C/R24C mice with mice deficient for p27 (but not with those deficient for p18) generates mice that develop pituitary adenomas with higher incidence and shorter latency than compound Cdk4 knock-in mice (Sotillo et al. 2005). Moreover, a dramatic cooperation in pituitary tumor development has been observed in mutant mice carrying a combination of the Cdk4 R24C, p21-null, and p27-null alleles (Quereda & Malumbres 2009).
Another protein involved in cell cycle regulation at different stages, and critical in pituitary alterations in mice, is PTTG1/securin. Indeed, transgenic mice overexpressing Pttg1 in the pituitary develop pituitary hyperplasia and focal microadenomas. Moreover, their crossing with pRb+/− mice led to the development of AP adenomas (Donangelo et al. 2006), whereas Pttg1 deletion results in pituitary p21 induction and abrogates tumor development in Rb+/− mice (Chesnokova et al. 2008).
Finally, mice overexpressing either members of the Hmga family (Hmga1 and Hmga2), or the truncated mutant Hmga2 (Battista et al. 1999), develop large and hypervascular AP adenomas (Fedele et al. 2002, 2005). These proteins play key roles in a wide range of biological processes including regulation of cell cycle (see below).
The HMGA proteins
The HMGA protein family includes HMGA1a and HMGA1b, which are encoded by the HMGA1 gene through an alternative splicing (Johnson et al. 1989), and the closely related HMGA2 protein (Manfioletti et al. 1991).
The HMGA1a, HMGA1b, and HMGA2 proteins are composed of 107, 96, and 108 amino acid residues respectively. Each of them contains three basic domains, named AT-hooks, through which they bind DNA in AT-rich regions inside the minor groove (Elton et al. 1987, Reeves & Nissen 1990), and an acidic carboxy-terminal region, which contains several hydroxylic amino acids that are phosphorylated by casein kinase II. These proteins play key roles in chromatin architecture and gene expression control by serving as generalized chromatin modifiers, either enhancing or suppressing the ability of several transcriptional factors to act on transcriptional regulation (Thanos & Maniatis 1995).
Both HMGA genes are widely expressed during embryogenesis, whereas they are absent or low expressed in adult cells and tissues (Zhou et al. 1995, Chiappetta et al. 1996, Rommel et al. 1997, Anand & Chada 2000). Recently, it has been shown that HMGA2 is highly expressed in neural fetal and young adult stem cells, being required for its self-renewal, relative to old adult stem cells, by negatively regulating p16In4a and p19Arf expression in fetal and young adult, but not in old adult, stages (Nishino et al. 2008).
HMGA overexpression represents a common feature of malignant neoplasias and a poor prognostic index since it often correlates with the presence of metastases and with a reduced survival (Fusco & Fedele 2007). The HMGA genes have a critical role also in the development of benign tumors of mesenchymal origin including lipomas. In fact, rearrangements of the HMGA2 gene, due to 12q13–15 chromosomal translocations, have been frequently detected in these tumors (Ashar et al. 1995, Schoenmakers et al. 1995). In most of these cases, the HMGA2 modifications comprise truncation of the gene with the loss of the sequences coding for the C-tail and the 3′-UTR regulatory region that contains multiple target sites for let-7, one of the founding members of the microRNA (miR) family (Lee & Dutta 2007, Mayr et al. 2007). Therefore, this truncation would result in HMGA2 overexpression due to the lack of the inhibitory effect by let-7.
As far as HMGA1 is concerned, chromosomal breakpoints causing rearrangements have not been found intragenically but are found either upstream or downstream of the HMGA1 gene, and sometimes within an 80 kb surrounding region (Kazmierczak et al. 1996, 1998).
Different studies, both in vitro and in vivo, have demonstrated that overexpression of the HMGA proteins is a necessary event in cell transformation: a) the block of HMGA1 protein synthesis by an antisense methodology prevents rat thyroid malignant cell transformation by acute murine retroviruses (Berlingieri et al. 1995, 2002); b) a recombinant adenovirus carrying HMGA1 sequences in an antisense orientation, and able to suppress HMGA1 protein synthesis, induces cell death in two human thyroid anaplastic carcinoma cell lines, but not in normal thyroid cells (Scala et al. 2000); c) increased expression of both HMGA1 and HMGA2 proteins leads to neoplastic transformation of Rat1a fibroblasts with anchorage-independent cell growth (Wood et al. 2000); d) shRNA-mediated HMGA1 silencing on lung and pancreatic carcinomas resulted in significant reductions in anchorage-independent proliferation in soft agar (Liau et al. 2007); and e) transgenic mice overexpressing either the Hmga1 or Hmga2 gene, under the transcriptional control of different promoters, develop several neoplasias including abdominal/pelvic lipomatosis, lipomas, mixed GH/PRL-secreting pituitary adenomas (Battista et al. 1999, Arlotta et al. 2000, Fedele et al. 2002), fibroadenomas of the breast, salivary gland adenomas (Zaidi et al. 2006), and mature T-cell lymphomas (Baldassarre et al. 2001, Xu et al. 2004).
HMGA proteins in pituitary tumorigenesis
Several pieces of evidence support a critical role of HMGA2 overexpression in the development of pituitary adenomas, particularly in prolactinomas that represent the most frequent histotype. Indeed, by dual-color interphase fluorescence in situ hybridization analysis using HMGA2-specific PACs and BACs, it was found that the HMGA2 locus was amplified in seven of the eight prolactinoma samples examined. The cytogenetic manifestations of elevated HMGA2 concentrations ranged from simple trisomy to tetrasomy of 12 and der(12) chromosomes to marker chromosomes bearing 12q14–15-derived regions (where the HMGA2 gene is located). Reverse transcription PCR, western blot, and immunohistochemical analysis showed HMGA2 overexpression in prolactinomas bearing rearrangement of regions 12q14–15 (Finelli et al. 2002). Conversely, the role of HMGA2 in development of NFPA is less evident since even though HMGA2 overexpression was observed in 12 out of 18 samples analyzed, the upregulation of the gene could be associated with amplification and/or rearrangement of the HMGA2 locus only in two cases (Pierantoni et al. 2005). Recently, HMGA2 expression has been analyzed using immunohistochemistry with respect to various clinicopathologic factors in 98 pituitary adenomas (Qian et al. 2009). Overexpression of HMGA2 was observed in 39% of pituitary adenomas compared with normal adenohypophyseal tissue, and it was frequently found in PRL-, ACTH-, or FSH/LH-secreting adenomas and in NFPA, but relatively rare in GH- and mixed GH/PRL-secreting adenomas. HMGA2 expression was significantly associated with tumor invasion and was significantly higher in grade IV than in grades I, II, and III adenomas (Qian et al. 2009).
The amplification of the HMGA2 locus mainly accounts for HMGA2 overexpression at least in prolactinomas. However, it is likely that increased dosage of chromosome 12 is insufficient to drive up activation of the HMGA2 gene, since it was found in NFPAs either expressing or not expressing the HMGA2 gene. Recent studies support the idea that decreased expression of miRs, able to modulate HMGA protein levels, may partially account for the abundant HMGA2 protein levels in pituitary adenomas. Indeed, decreased expression of let-7 was observed in 23 of 55 (42%) adenomas with an inverse correlation between let-7 and HMGA2 expressions (Qian et al. 2009). Moreover, it has been shown that miRs 15, 16, and 196, which target the HMGA mRNAs (De Martino et al. 2009a, Kaddar et al. 2009, Palmieri et al. manuscript in preparation), are downregulated in human prolactinomas (Bottoni et al. 2005, Palmieri et al. manuscript in preparation).
The critical role of HMGA2 protein in pituitary oncogenesis is validated by the phenotype of transgenic mice overexpressing a truncated or a wild-type Hmga2 gene under the transcriptional control of the strong and ubiquitous CMV promoter (Fedele et al. 2002). In fact, most of these mice (85% of females and 40% of males) showed the onset of pituitary adenomas secreting PRL and GH (Fedele et al. 2002).
Differently from HMGA2, the causal role of HMGA1 gene in pituitary carcinogenesis is still to be defined. In fact, even though both HMGA1 and HMGA2 transcripts have been found upregulated in a panel of 45 human pituitary adenomas (De Martino et al. 2009b), and transgenic mice overexpressing Hmga1 develop pituitary adenomas secreting PRL and GH (Fedele et al. 2005), no rearrangements of the HMGA1 locus have been found. Moreover, the detection of HMGA1 overexpression in all pituitary adenomas subtypes seems to indicate that HMGA1 induction may represent a secondary event in the tumorigenesis of pituitary gland.
HMGA2 and the pituitary cell cycle
Recent pieces of evidence indicate that HMGA2 protein is involved in the development of pituitary adenomas by interfering with the cell cycle machinery. Indeed, it has been demonstrated in the mouse model of Hmga2 transgenic mice that the positive role of HMGA2 in pituitary cell proliferation is due to the interaction with the RB-E2F pathway, suggesting a new class of cell cycle-related proteins: the suppressors of the cell cycle inhibitors (Fedele et al. 2006). In more detail, HMGA2 binds to pRB A/B pocket domain thereby displacing the HDAC1 protein from pRB. This displacement results in the recruitment of HDAC to the E2F1 target promoters and acetylation of both histones and other proteins including E2F1. The acetylation of histones opens up the chromatin and facilitates gene transcription. Moreover, acetylation of E2F1 augments its DNA binding and stabilizes the protein in its free active form, thereby enhancing the expression of the E2F target genes, such as CDC6 and TK1 that promote the G1/S transition. The crucial role of enhanced E2F1 activity in the onset and progression of pituitary adenomas was validated by the rescue of the adenomatous phenotype when transgenic mice overexpressing the Hmga2 gene were mated with E2f1 knockout mice. In fact, the majority of double mutant mice did not develop tumors, and only a few of them showed small and slow growing adenomas (Fedele et al. 2006).
The alteration of cell cycle as a critical event in the induction of pituitary adenomas by the HMGA2 gene seems further confirmed by the direct role of HMGA proteins in the upregulation of cyclin B2 gene transcription, leading to overexpression of cyclin B2 in pituitary adenomas developed by mice carrying Hmga transgenes (De Martino et al. 2009b). Consistently, an increased expression of CCNB2, the human gene coding for the cyclin B2 protein, directly correlates with HMGA1 and HMGA2 expression in human pituitary adenomas of different histotypes (De Martino et al. 2009b). Since cyclin B2, complexed with CDK1, plays a critical role in regulating the G2/M phase transition of the cell cycle, it is reasonable to hypothesize that cyclin B2 induction by HMGA proteins may contribute to increase cell proliferation of the pituitary cells where HMGA proteins are overexpressed. Interestingly, the analysis of the mRNA expression profile of the pituitary adenomas arising in Hmga transgenic mice also revealed the overexpression of other genes, such as Ccnb1, Ccnd3, and Cdc2, coding for proteins involved in cell cycle regulation (De Martino et al. 2007). It is likely that their altered expression may have a role in experimental and human pituitary neoplasias. However, further studies are required to define the role of these proteins in pituitary tumorigenesis.
Concluding remarks
In conclusion, these recent findings provide new insight with respect to the mechanisms that dysregulate the cell cycle in pituitary tumorigenesis. By acting as regulators of different steps of cell cycle progression, the HMGA proteins play a crucial role in the maintenance of the pituitary cell homeostasis. In fact, overexpression of HMGA proteins in mice is sufficient to get a pituitary phenotype only comparable to that observed by the cooperation of two genetic alterations affecting different cell cycle regulators. Also, it is possible that other mechanisms may contribute to the role of HMGA2 in pituitary tumorigenesis, consistent with the ability of HMGA proteins to either activate or repress many different genes.
Since the role of the HMGA2 overexpression represents a critical event in the development of a significant number of prolactinomas, a therapy based on the suppression of the HMGA2 function may be envisaged for the treatment of recurrent prolactinomas. However, as cell cycle alterations can arise independently from the event that has caused them, and they are crucial in pituitary tumorigenesis, therapies aimed to target cell cycle actors may be also conceived. Even not taking into account of the role of HMGA proteins in pituitary cell cycle, transgenic mice overexpressing either Hmga1 or Hmga2 may be considered excellent animal models to explore new therapies for pituitary adenomas (Fedele et al. 2007).
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
The authors are supported by grants from Associazione Italiana Ricerca sul Cancro (AIRC) and Programmi di Ricerca Scientifica di Rilevante Interesse Nazionale (PRIN).
Acknowledgements
M Fedele thanks R Visone, I De Martino, and D Palmieri for their outstanding contribute, during the years, on pituitary work.
- Revision received 25 February 2010
- Accepted 10 March 2010
- © 2010 Society for Endocrinology










