- Made available online as an Accepted Preprint 22 July 2009
- Accepted Preprint first posted online on 22 July 2009
AMPK as a mediator of hormonal signalling
- Endocrinology, Barts and the London School of Medicine and Dentistry, William Harvey Research Institute, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK
- (Correspondence should be addressed to M Korbonits; Email: m.korbonits{at}qmul.ac.uk)
Abstract
AMP-activated protein kinase (AMPK) is a key molecular player in energy homeostasis at both cellular and whole-body levels. AMPK has been shown to mediate the metabolic effects of hormones such as leptin, ghrelin, adiponectin, glucocorticoids and insulin as well as cannabinoids. Generally, activated AMPK stimulates catabolic pathways (glycolysis, fatty acid oxidation and mitochondrial biogenesis) and inhibits anabolic pathways (gluconeogenesis, glycogen, fatty acid and protein synthesis), and has a direct appetite-regulating effect in the hypothalamus. Drugs that activate AMPK, namely metformin and thiazolidinediones, are often used to treat metabolic disorders. Thus, AMPK is now recognised as a potential target for the treatment of obesity and associated co-morbidities.
Introduction
AMP-activated protein kinase (AMPK) has emerged as a key molecular player in energy homeostasis at both cellular and whole-body levels (Kahn et al. 2005). Initially, AMPK was shown to have lipid-related effects: it inactivates acetyl-CoA carboxylase (ACC; Carlson & Kim 1973) and 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase (Beg et al. 1973), the key regulatory enzymes of fatty acid and cholesterol synthesis. Later, the role of AMPK in carbohydrate and protein metabolism, cell cycle regulation and mitochondrial biogenesis was also described. AMPK is an evolutionarily conserved serine/threonine kinase with a catalytic α-subunit and regulatory β- and γ-subunits, forming a heterotrimeric complex. The upstream regulation of AMPK is summarised in Figs 1 and 2.
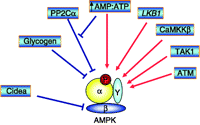
AMPK regulation: AMPK's upstream kinases phosphorylate the Thr172 residue of the α-kinase subunit. LKB1, a tumour-suppressor gene (Hawley et al. 2003, Woods et al. 2003, Shaw et al. 2004), calmodulin kinase kinase (CaMKK; Hawley et al. 2005, Hurley et al. 2005, Woods et al. 2005), and the recently described kinases transforming growth factor-β-activated kinase (TAK1) and ataxia telangiectasia mutated (ATM) are the known AMPK kinases. CaMKK2 (also known as CaMKKβ) is expressed primarily in the brain, suggesting that this Ca2+-mediated pathway may be operating in neurons (Towler & Hardie 2007). TAK1 is a member of the mitogen-activated protein kinase kinase kinase family and is reported to phosphorylate AMPK in HeLa cells, which lack LKB1 expression (Momcilovic et al. 2006). ATM, which stimulates mitochondrial biogenesis in response to double-stranded DNA breaks, also can phosphorylate AMPK (Fu et al. 2008). A rise in AMP:ATP ratio activates AMPK i) allosterically and ii) by inhibition of dephosphorylation by protein phosphatases (Hardie et al. 1999, Sanders et al. 2007). Recently, it has been shown that AMPK can be regulated directly by glycogen and may act as a glycogen sensor (McBride et al. 2009). The α1–6 linkage-branched forms of glycogen allosterically inhibit AMPK more potently than the linear α1–4-linked forms (Carling 2009, McBride et al. 2009). In addition to the allosteric effect, the branched glycogen also inhibits LKB1- or CaMKK2-induced Thr172 phosphorylation, while dephosphorylation is not affected by glycogen. A new model of AMPK regulation, which is independent of AMP, has recently been identified. It is shown that cell death-inducing DFFA-like effector-a (Cidea) forms a complex with the β-subunit of AMPK, eliciting a ubiquitination-mediated degradation of AMPK (Qi et al. 2008).
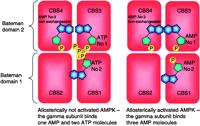
The γ-subunit of AMPK contains three AMP-binding sites. These are formed in the interface of its two pairs of CBS domains, also called Bateman domains. Two of these binding sites can bind either AMP or ATP, whereas a third site contains a tightly bound AMP that does not exchange (Xiao et al. 2007). When AMPK is inactive under physiological conditions, it binds two ATP and one AMP molecule, while in low energy states it binds three AMP molecules. It is now proposed that the interaction of the catalytically active kinase domain with the AMP-bound γ-subunit protects the phosphorylated Thr172 residue from dephosphorylation.
Role of AMPK in the central control of appetite
AMPK is expressed throughout the brain: all isoforms are expressed in neuronal tissues including areas that are involved in the control of food intake and neuroendocrine function, such as the hypothalamus and the hindbrain (Turnley et al. 1999, Kola 2008). In the last 5 years, AMPK has emerged as a nutrient and glucose sensor in the hypothalamus (Momcilovic et al. 2006). Hypothalamic AMPK activity is increased during fasting and decreased during refeeding (Minokoshi et al. 2004). Pharmacological activation of AMPK in the rodent hypothalamus with 5-aminoimidazole-4-carboxamide riboside (AICAR) causes an increase in food intake (Xue & Kahn 2006). Alteration in ventromedial hypothalamic AMPK activity with recombinant adenoviruses expressing dominant negative (DN) or constitutively active (CA) AMPK-α1/α2 subunit (Minokoshi et al. 2004) changed body weight and food intake. DN-AMPK adenovirus-treated mice ate less and had lower body weight compared with control mice. DN-AMPK mice also had decreased neuropeptide Y (NPY) and agouti-related peptide (AgRP) mRNA levels in the arcuate nucleus (ARC) under ad libitum fed conditions. In contrast, CA-AMPK adenovirus-treated mice ate significantly more and had higher body weight with increased expression of NPY and AgRP mRNA in ARC, as well as increased orexigenic melanin-concentrating hormone expression in lateral hypothalamus. This suggests that high AMPK activity enhances orexigenic signals, whereas low AMPK activity suppresses these signals under ad libitum fed conditions. In agreement with this, deletion of AMPKα2 in AgRP neurons led to the development of an age-dependent lean phenotype.
Peripheral hormones from the gastrointestinal tract (peptide YY, ghrelin, cholecystokinin, glucagon-like peptide 1 (GLP-1) and oxyntomodulin) and adipose tissue (leptin, resistin and adiponectin) are important in influencing the activity of the appetite-regulating neuronal populations in the hypothalamus. In addition, a number of these hormones have been shown to influence AMPK activity. In the short term, anorectic agents such as glucose, GLP-1 and oxyntomodulin decrease hypothalamic AMPK activity (Andersson et al. 2004, Minokoshi et al. 2004, Seo et al. 2008), leading to reduction in food intake during satiation, while orexigenic agents such as ghrelin lead to AMPK activation and increased food intake (Andersson et al. 2004, Kola et al. 2005). In the long term, the circulating anorectic insulin and leptin determine the energy and adiposity profile.
The hypothalamus is not the only location in the brain important for appetite regulation. Emerging data suggest that the nucleus tractus solitarius (NTS) in the hindbrain also has an important role. Fasting increases AMPK activity in the NTS and leptin inhibits it (Hayes et al. 2009a). Ghrelin is known to activate neurons in the NTS (Date et al. 2006). GLP-1 (7–36) amide, an anorectic hormone, acts both in the hypothalamus and the NTS (Goldstone et al. 2000, Seo et al. 2008, Hayes et al. 2009b).
Role of AMPK in peripheral tissues
AMPK is ubiquitously expressed and plays an important role in the peripheral metabolism of the skeletal muscle, liver, fat, myocardium and other tissues. In general, activated AMPK switches on catabolic processes that produce ATP and switches off ATP-consuming processes, thus restoring the AMP:ATP ratio.
AMPK plays a key role in regulating lipid metabolism. Activated AMPK phosphorylates and inhibits ACC1 and HMG-CoA, decreases fatty acid synthase (FAS) expression and activates malonyl-CoA carboxylase, thereby leading to a decrease in fatty acid and cholesterol synthesis (Woods et al. 2000, Kahn et al. 2005, Lopez et al. 2007). Activated AMPK stimulates fatty acid oxidation by decreasing malonyl-CoA levels through the inhibition of ACC2 (Merrill et al. 1997, Kahn et al. 2005, Lopez et al. 2007). This leads to an increase in carnitine palmitoyltransferase 1 (CPT1) activity and the subsequent activation of fatty acid oxidation (Kahn et al. 2005, Lopez et al. 2007). The decreased AMPK activity in visceral fat could enhance lipolysis as well as lipogenesis, although the effect on lipogenesis prevails (Divertie et al. 1991, Djurhuus et al. 2002). AMPK thus plays a key role in regulating lipid metabolism. AMPK has been suggested to inhibit catecholamine-stimulated lipolysis in adipocytes (Corton et al. 1995, Daval et al. 2005), thus lowering the plasma level of fatty acids. Activated AMPK also stimulates and upregulates the expression of peroxisome proliferator-activated receptor-γ coactivator-1α, which consequently increases mitochondrial biogenesis (Terada et al. 2002, Zong et al. 2002).
AMPK also regulates glucose homeostasis. Activation of AMPK by contraction in fast-twitching muscles increases hexokinase II expression (Holmes et al. 1999), and enhances glucose uptake through the translocation of glucose transporter 4 (GLUT4) to the cell membrane and the upregulation of Glut4 gene expression (Holmes et al. 1999, Derave et al. 2000, Wright et al. 2005). Interestingly, these effects were not observed in slow-twitching soleus muscle (Derave et al. 2000, Wright et al. 2005).
AMPK regulates hepatic gluconeogenesis by inhibiting the transcription of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase; Lochhead et al. 2000, Cool et al. 2006). AMPKα2-knockout (KO) and LKB1-KO mice were shown to have glucose intolerance and fasting-induced hyperglycaemia, possibly caused by increased gluconeogenesis associated with increased PEPCK and G6Pase activity (Lochhead et al. 2000, Cool et al. 2006). Activated AMPK in skeletal muscle phosphorylates and inhibits glycogen synthase, thereby leading to a decrease in glycogen synthesis (Wojtaszewski et al. 2002).
Hypothalamic AMPK has been linked to the regulation of peripheral metabolism, suggesting that AMPK is a key enzyme in coordinating the interaction between peripheral and central energy regulation. Central AICAR treatment has been shown to increase both insulin-mediated and non-insulin-mediated glycogen synthesis (Perrin et al. 2004), thus implicating the role of hypothalamic AMPK in regulating muscle glycogen synthesis. Central insulin infusion also increased muscle glycogen synthesis and this effect was blocked by the co-administration of glucose, possibly mediated by AMPK (Perrin et al. 2004). Central adiponectin treatment leads to hypothalamic AMPK activation and decreases energy expenditure, possibly via a reduced expression of uncoupling protein-1 (UCP-1) in brown adipose tissue (Kubota et al. 2007). Central α-lipoic acid, which inhibits hypothalamic AMPK activity, increases UCP-1 expression and energy expenditure in brown adipose tissue (Kim et al. 2004b). Central ghrelin treatment, independently from the effect on food intake, has been shown to increase glucose utilisation rate of white and brown adipose tissues and counteract the effects of intracerebroventricular leptin treatment on fat weight, plasma glucose and insulin (Kim et al. 2004a, Theander-Carrillo et al. 2006).
Role of AMPK as mediator of hormonal signals
Intriguingly, several hormones have tissue-specific, often opposite, effects on AMPK activity (Fig. 3).
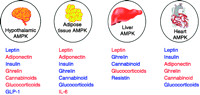
Hormones that activate (red) or inhibit (blue) AMPK in various tissues.
Leptin
Leptin increases AMPK activity in the skeletal muscle directly as well as indirectly through stimulation of the hypothalamo-sympathetic axis (Minokoshi et al. 2002). Chronic s.c. administration of leptin also increases the expression of AMPK in skeletal muscle (Steinberg et al. 2003). Leptin- or leptin receptor-deficient rodents showed a decreased AMPK activity in the liver (Yu et al. 2004). In lean animals, leptin has been shown to attenuate hepatic glucose production and insulin resistance under normal conditions and to slightly increase AMPK activity (Brabant et al. 2005). However, these effects are lost in diet-induced obese rats, thereby suggesting an important physiological dysregulation of leptin effects in obese animals. Leptin inhibits triacylglycerol storage and stimulates fatty acid oxidation in the heart, and both AMPK-dependent (Lee et al. 2004) and AMPK-independent (Atkinson et al. 2002) pathways have been suggested.
Central injection of leptin into ventromedial hypothalamus (VMH) of rats has been shown to increase glucose uptake in the heart, brown adipose tissue and skeletal muscle, but not in white adipose tissue (Kamohara et al. 1997, Haque et al. 1999, Minokoshi et al. 1999). Central infusion of leptin decreases hepatic glycogen content (Kamohara et al. 1997, Haque et al. 1999).
In the hypothalamus, leptin has an opposite effect: it decreases AMPK activity in the ARC and paraventricular (PVC) nuclei (Minokoshi et al. 2002, 2004, Andersson et al. 2004, Mountjoy et al. 2007). By reducing the appetite centrally and increasing the peripheral fatty acid consumption, these tissue-specific effects of leptin lead to an overall negative energy balance and reduction in body weight.
Adiponectin
Adiponectin activates and stimulates liver and muscle AMPK activity in vivo and in vitro, leading to stimulation of glucose uptake, fatty acid oxidation and PEPCK (Yamauchi et al. 2002). These lead to an improvement in insulin sensitivity. Globular adiponectin was also shown to activate AMPK in primary rat adipocytes (Tomas et al. 2002, Yamauchi et al. 2002, Wu et al. 2003, Huypens et al. 2005).
Adiponectin protects the heart from ischaemic injury via AMPK- and cyclooxgenase-2-dependent mechanisms (Shibata et al. 2005). Adiponectin is also suggested to play a beneficial role in cardiac remodelling through multiple mechanisms, one of which is possibly via the activation of AMPK (Liao et al. 2005, Shibata et al. 2005). Impaired regulation of AMPK and glucose metabolism in adiponectin-deficient mice result in the development of heart failure (Liao et al. 2005). In endothelial cells, adiponectin stimulates nitric oxide production via AMPK activation, leading to beneficial vasoprotective effects (Chen et al. 2003).
It has been suggested that adiponectin may be involved in the stimulation of food intake (Kadowaki et al. 2008). Serum and cerebrospinal fluid (CSF) adiponectin concentrations increase under fasting conditions, as does expression of adiponectin receptor-1 in ARC. Central adiponectin administration leads to increased phosphorylation of AMPK and ACC in the hypothalamus (Carling 2005, Xue & Kahn 2006, Kadowaki et al. 2008). Adiponectin KO mice were shown to have reduced food intake and decreased AMPK activity in ARC (Kadowaki et al. 2008). Thus, unlike leptin, adiponectin stimulates both central and peripheral AMPK activity. Adiponectin-transgenic ob/ob mice, which have serum adiponectin levels two- to threefold higher than ob/ob mice, have significantly higher body weight, but an improved metabolic state compared with ob/ob mice (Kim et al. 2007). Adiponectin is considered a starvation hormone: under fasting conditions, high adiponectin levels stimulate central and peripheral AMPK leading to increased food intake and decreased energy expenditure, promoting fat storage. After refeeding, adiponectin levels would fall with a consequent decrease in AMPK activity leading to reduced food intake and an increase in energy expenditure.
Resistin
Resistin is an adipokine secreted in rodents and humans, which generally seems to have opposite effects to those of adiponectin. Resistin induces insulin resistance and stimulates hepatic glucose production. These effects are thought to be mediated by a reduction in liver AMPK activity (Banerjee et al. 2004, Muse et al. 2004). It has been shown to decrease fatty acid uptake and oxidation in skeletal muscle via a reduction in the membrane content of fatty acid translocase/CD36, possibly mediated by inhibition of AMPK (Palanivel & Sweeney 2005). Resistin, despite its anorectic effect, has been shown to phosphorylate hypothalamic AMPK and ACC. The consequent inactivation of ACC, after AMPK activation, might represent a physiological compensatory mechanism that prevents deleteriously high levels of malonyl-CoA occurring in the hypothalamus after resistin-induced FAS inhibition in the VMH (Vazquez et al. 2008). Time course-dependent resistin treatments would probably be needed to clarify this issue.
Ghrelin and cannabinoids
Ghrelin has been shown to regulate AMPK activity in hypothalamus and peripheral tissues (Andersson et al. 2004, Kola et al. 2005). Cannabinoids are known to regulate appetite and peripheral metabolism, and AMPK has been shown to mediate these effects (Kola et al. 2005, 2008). Both ghrelin and cannabinoids have similar effects on AMPK activity in various tissues: they stimulate hypothalamic and heart AMPK activity, while inhibit adipose tissue and liver AMPK activity (Kola et al. 2005, 2008).
The mechanism of central effect of ghrelin includes the activation of Ca2+ signalling in NPY neurons in the ARC (Kohno et al. 2003, 2008). The Ca2+ rise leads to CAMKK2 activation, which can stimulate hypothalamic AMPK (Anderson et al. 2008, Sleeman & Latres 2008). AMPK activation leads to inhibition of malonyl-CoA and stimulation of CPT1, leading to increased mitochondrial oxidation and activation of the UCP-2, which can increase NPY/AgRP neuronal activity and ultimately stimulate appetite (Andrews et al. 2008, Lopez et al. 2008).
Endocannabinoids are synthesised locally on demand, and the level in the hypothalamus varies in response to feeding and fasting (Kirkham et al. 2002). The variation in hypothalamic endocannabinoid levels seems to play an important role in mediating the anorectic effects of leptin (Di Marzo et al. 2001) and the orexigenic effects of ghrelin (Kola et al. 2008). Recently, we have shown that the effects of ghrelin on hypothalamic AMPK activity and appetite are abolished in the absence of cannabinoid type 1 receptor (CB1) or in the presence of a CB1 antagonist rimonabant (Kola et al. 2008). These data suggest that an intact cannabinoid-signalling pathway is required for the effects of ghrelin on AMPK activity and appetite. Interestingly, i.p. injection of cannabinoids in rats results in increased plasma ghrelin levels (Zbucki et al. 2008). This suggests that the stimulation of appetite by cannabinoids may be connected to an increase in ghrelin secretion from the gastric X/A-like cells (Zbucki et al. 2008). Further studies are needed to elucidate the details of the ghrelin–cannabinoid interaction.
Insulin
Insulin has a range of metabolic effects in addition to its main role of stimulating glucose uptake into the cells. Centrally, insulin is an anorectic hormone, which has been shown to inhibit hypothalamic AMPK activity (Minokoshi et al. 2004). Insulin deficiency has been proposed as one of the factors causing hypothalamic AMPK activation and the subsequent increase in food intake seen in streptozotocin-induced diabetic rats (Namkoong et al. 2005). In the periphery, insulin inhibits AMPK in fat by activating protein kinase B/Akt complex, which can phosphorylate alphaAMPK at Ser485/491, thus leading to reduced phosphorylation at Thr172 (Kovacic et al. 2003, Horman et al. 2006). Insulin inhibits myocardial AMPK activity during ischaemic events alone or when co-administered with glucose, suggesting that the inhibitory effect of insulin on myocardial AMPK activity might be caused by enhanced glucose metabolism (Russell et al. 2004).
Insulin resistance is characterised by the inability of insulin to increase glucose uptake and repress glucose production in the liver, and it often leads to hyperglycaemia. The role of AMPK in this condition has to be considered. While activated AMPK stimulates catabolic pathways and inhibits the energy-consuming anabolic processes, insulin promotes glycogen, lipid and protein synthesis. However, both upregulate glucose uptake in muscle via an effect of GLUT1 or GLUT4 translocation and increase in GLUT4 transcription (Zheng et al. 2001, Barnes et al. 2002). In skeletal muscle, the two pathways also phosphorylate the protein AS160, which has a Rab GTPase-activating protein domain that can increase translocation of GLUT4 to the plasma membrane (Kurth-Kraczek et al. 1999, Kramer et al. 2006, Treebak et al. 2006). These factors will lead to an increase in glucose uptake, which is an important homeostatic feature of plasma glucose regulation. Furthermore, AMPK activation is thought to upregulate insulin receptor substrate-1 (Harrington et al. 2004, Shah et al. 2004, Um et al. 2004) through inhibition of the insulin–mammalian target of rapamycin (mTOR) pathway (Fisher et al. 2002, Inoki et al. 2003). This will improve the insulin sensitivity profile. Both insulin and activated AMPK repress the expression of the gluconeogenic enzymes PEPCK and G6Pase (Lochhead et al. 2000). Phosphorylation of AMPK is thought to result in translocation of the transcriptional coactivator TORC2 to the cytoplasm (Koo et al. 2005), thus repressing the expression of the TORC2-target enzymes
Glucagon-like peptide-1
GLP-1 is produced from pre-proglucagon mRNA, which is expressed in NTS cell bodies in the brainstem, with projections to the PVN and other hypothalamic nuclei involved in the control of feeding (Goldstone et al. 2000). Central nervous system GLP-1 is an endogenous inhibitor of feeding acting via the GLP-1 receptor. Hypothalamic GLP-1 peptide content is decreased during fasting. Fasting-induced increase in hypothalamic AMPK activity is inhibited by GLP-1 and this could be the mechanism of its anorectic effects (Seo et al. 2008). It has been shown that the anorectic effects of leptin are at least partly via GLP-1. GLP-1 acts in both the hypothalamus and the NTS, as GLP-1 receptors located in the NTS are also suggested to regulate food intake (Hayes et al. 2009b).
Glucocorticoids
Glucocorticoids increase appetite and lead to increased availability of metabolic fuels such as amino acids and fatty acids. Chronic or excessive exposure to glucocorticoids will result in insulin resistance, truncal obesity, hyperlipidaemia and symptoms similar to the metabolic syndrome. We have suggested that AMPK is involved in the central and peripheral effects of glucocorticoids. Glucocorticoids activate hypothalamic AMPK activity in vivo (Christ-Crain et al. 2008) either directly or via stimulation of endocannabinoid synthesis (Di et al. 2005, Christ-Crain et al. 2008), and these effects could lead to appetite stimulation (Tataranni et al. 1996). In the periphery, glucocorticoids inhibit AMPK activity in adipose tissue, leading to increased lipogenesis and fat storage (Christ-Crain et al. 2008). Glucocorticoids also inhibited AMPK activity in the heart, which might, at least in part, mediate the detrimental effects of glucocorticoid excess on the heart. Surprisingly, and somewhat unexpectedly, glucocorticoids were shown to stimulate AMPK activity in rat liver in vivo as well as in a liver cell line (Viana et al. 2006, Christ-Crain et al. 2008). This could be the result of the balance of local lipolysis, lipid oxidation and the flux of fatty acids into the liver (Foretz et al. 2005). It has been shown that an increase in free fatty acids leads to fatty acid esterification, an energy-demanding process, and this can increase the cellular AMP:ATP ratio and therefore AMPK activity (Gauthier et al. 2008).
Inflammatory mediators and AMPK
Interleukin-6 (IL-6) treatment was shown to increase AMPK phosphorylation in cultured rodent myocytes and adipocytes, as well as in muscle, liver and adipose tissue in vivo (Keller et al. 2001, Park et al. 2002, Kahn et al. 2005). Moreover, IL-6-KO mice have decreased AMPK activity in muscle (Kelly et al. 2004). Conversely, adiponectin was shown to increase IL-6 production in human synovial fibroblasts partly via AMPK regulation (Tang et al. 2007). Exercise increases AMPK activity in skeletal muscle primarily in response to changes in the AMP:ATP ratio (Ruderman et al. 2006). Exercise affects AMPK activity in fat and liver tissue (Takekoshi et al. 2006) possibly due to an increase in circulating levels of IL-6 (Keller et al. 2001). Furthermore, AMPK activity is often still increased after exercise at times when the energy state of the muscle is presumably no longer altered, and IL-6 is suggested to be involved in AMPK activation during this ‘late-phase’ stage (Ruderman et al. 2006).
Ciliary neurotrophic factor (CNTF) is a cytokine, which was found to induce severe anorectic effect (Miller et al. 1996), possibly via hypothalamic neurogenesis that leads to sustained reduction in caloric intake and prolonged maintenance of weight loss (Kokoeva et al. 2005). CNTF was shown to activate AMPK through the CNTFRα-IL-6R-gp130β receptor complex and ultimately increasing fatty acid oxidation and reducing insulin resistance in skeletal muscle (Steinberg et al. 2006b). CNTF can also suppress inflammatory signalling cascades associated with lipid accumulation in the liver and skeletal muscle (Febbraio 2007).
The ischaemic heart releases macrophage inhibitory factor (MIF), an upstream regulator of inflammation. MIF stimulates AMPK through CD74 during ischaemia, and shows impaired ischaemic AMPK signalling in the heart of mice with germline deletion of the MIF gene (Miller et al. 2008). MIF promotes glucose uptake and protects the heart during ischaemia–reperfusion injury (Miller et al. 2008). Human fibroblasts with a low-activity MIF promoter polymorphism also have diminished MIF release and AMPK activation during hypoxia, thus linking inflammation with metabolism in the heart (Miller et al. 2008).
Tumour necrosis factor α (TNFα) signalling via TNF-receptor has been shown to suppress skeletal muscle AMPK activity both in vivo and in vitro (Steinberg et al. 2006a). This happens via upregulation of protein phosphatase 2C transcription, which in turn reduces ACC phosphorylation and fatty acid oxidation as well as increasing diacylglycerol accumulation in the muscle. Suppressive effects of TNFα on AMPK activity, seen in obese mice with pathologically elevated levels of TNFα, could be reversed in null mice for both TNF receptor-1 and -2 or following treatment with a TNFα-neutralising antibody (Steinberg et al. 2006a). This indicates that AMPK is an important target for TNFα signalling.
Metabolic syndrome and AMPK
The metabolic abnormalities observed in metabolic syndrome are insulin resistance, hypertriglyceridaemia, abdominal obesity, hypertension, reduced levels of the beneficial high-density lipoprotein and disturbances in glucose metabolism (Trevisan et al. 1998). Patients with metabolic syndrome have higher risks of developing cardiovascular disease (Isomaa et al. 2001) and have higher rates of mortality from all causes (Trevisan et al. 1998). Downstream targets of AMPK such as genes regulating carbohydrate metabolism (e.g. glycogen synthetase, ChREBP) or lipid metabolism (e.g. HMG-CoA, FAS, ACC, SREBP1) play an important role in features of the metabolic syndrome. Therefore, AMPK emerged as a target for treatment of the metabolic syndrome. Two major anti-diabetic drugs that exert effects via the AMPK pathway will be considered in this review: metformin and rosiglitazone.
Metformin
Metformin, a biguanide agent, is widely used as an anti-diabetic drug. The biguanide class of anti-diabetic drugs originates from the French lilac (Galega officinalis) plant, known for several centuries to reduce the symptoms of diabetes mellitus (Witters 2001). These were first introduced in 1957 and marketed in France in 1979.
Metformin is shown to stimulate AMPK in the liver and in the muscle (Zhou et al. 2001, Zang et al. 2004, Shaw et al. 2005). This consequently stimulates glucose uptake in the muscle, induces hepatic fatty acid oxidation and inhibits hepatic glucose production and expression of lipogenic enzymes. Metformin does not activate AMPK directly, but indirectly via inhibition of complex I of the respiratory chain and the consequent increase in AMP:ATP ratio. LKB1 has also been reported to mediate the activation of AMPK in the liver by metformin (Zhou et al. 2001, Zang et al. 2004, Shaw et al. 2005).
In contrast, metformin inhibits AMPK in the hypothalamus (Chau-Van et al. 2007). Metformin inhibits low glucose-induced AMPK phosphorylation and NPY mRNA expression. These may explain the anorectic effects of metformin.
Metformin is shown to reduce mitochondrial ATP synthesis in the pancreatic β-cell, resulting in impaired glucose responsiveness, inhibition of insulin release and possibly apoptosis (Kefas et al. 2004, Leclerc et al. 2004). These findings of metformin are clearly undesired and further studies are needed to reassess the long-term effects of metformin on β-cells.
Rosiglitazone
Rosiglitazone belongs to a class of anti-diabetic drugs known as thiazolidinediones (TZDs). TZDs are used to reverse insulin resistance and improve glucose tolerance. It is known that TZDs improve insulin sensitivity by activating nuclear PPAR-γ and the consequent regulation of gene transcription. However, it is also believed that TZDs can improve insulin sensitivity via PPAR-γ-independent mechanisms, one of which is AMPK activation (Kahn et al. 2005).
Rosiglitazone was shown to increase AMPK activity in muscle cell lines (Fryer et al. 2002), and chronic rosiglitazone treatment was reported to restore skeletal muscle AMPKα2 activity in obese, insulin-resistant Zucker rats (Lessard et al. 2006). Rosiglitazone activates AMPK indirectly by inhibiting complex I of the respiratory chain, which consequently leads to an increase in cellular AMP:ATP ratio (El-Mir et al. 2000, Owen et al. 2000, Brunmair et al. 2004).
TZDs also decrease the levels of resistin and stimulate release of adiponectin via action on PPAR-γ in adipocytes (Samaha et al. 2006). These effects might also contribute to the stimulatory effect of TZDs on AMPK.
Conclusion
AMPK is one of the key regulators in energy homeostasis and is known to mediate the effects of several metabolic hormones. AMPK is now recognised as a potential target for the treatment of obesity and the metabolic syndrome.
Funding
C T Lim was supported by a Wellcome Trust Summer Studentship, and B Kola and M Korbonits are supported by a Wellcome Trust Research Grant.
Acknowledgements
We are very grateful to Dr Steve Gamblin (MRC, London) for the helpful comments on the structural description of AMPK activation.
- Revision received 26 June 2009
- Accepted 22 July 2009
- © 2010 Society for Endocrinology










