- Made available online as an Accepted Preprint 20 July 2009
- Accepted Preprint first posted online on 20 July 2009
Signal transduction of the CB1 cannabinoid receptor
- 1Department of Physiology, Faculty of Medicine, Semmelweis University, PO Box 259, H-1444 Budapest, Hungary
2Laboratory of Neurobiochemistry and Molecular Physiology, Semmelweis University and Hungarian Academy of Sciences, Budapest, Hungary
- (Correspondence should be addressed to L Hunyady at Department of Physiology, Faculty of Medicine, Semmelweis University; Email: hunyady{at}eok.sote.hu)
Abstract
The CB1 cannabinoid receptor (CB1R) is the major cannabinoid receptor in neuronal cells and the brain, but it also occurs in endocrine cells and other peripheral tissues. CB1R is a member of the superfamily of G-protein-coupled receptors (GPCRs), which are characterized by seven transmembrane helices. The major mediators of CB1R are the G proteins of the Gi/o family, which inhibit adenylyl cyclases in most tissues and cells, and regulate ion channels, including calcium and potassium ion channels. Regulation of ion channels is an important component of neurotransmission modulation by endogenous cannabinoid compounds released in response to depolarization and Ca2+-mobilizing hormones. However, evidence exists that CB1Rs can also stimulate adenylyl cyclase via Gs, induce receptor-mediated Ca2+ fluxes and stimulate phospholipases in some experimental models. Stimulation of CB1R also leads to phosphorylation and activation of mitogen-activated protein kinases (MAPK), such as p42/p44 MAPK, p38 MAPK and c-Jun N-terminal kinase, which can regulate nuclear transcription factors. Activated and phosphorylated CB1Rs also associate with β-arrestin molecules, which can induce the formation of signalling complexes and participate in the regulation of GPCR signalling. Recent data also suggest that CB1Rs can form homo- and heterodimers/oligomers, and the altered pharmacological properties of these receptor complexes may explain the pharmacological differences observed in various tissues.
Introduction
Marijuana or cannabis is a very popular recreational drug due to its ability to alter sensory perception and cause euphoria. However, it has also been recognized thousands of years ago that extracts of Cannabis sativa can exert medicinal effects (Pacher et al. 2006). The correct chemical structure of the main psychoactive ingredient of marijuana, Δ9-tetrahydrocannabinol (THC), was identified by Gaoni (1964). Although the lipophylic nature of cannabinoids slowed down the progress of pharmacological identification of its biological target, high-affinity-binding sites for cannabinoids in brain membranes were first reported in 1988 (Devane et al. 1988). Molecular biological studies have identified two major cannabinoid receptors, the CB1 receptor (CB1R) and CB2 receptor (CB2R; Matsuda et al. 1990, Munro et al. 1993). Anandamide and 2-arachidonylglycerol (2-AG) have been identified as the major endocannabinoids, but other endocannabinoid mediators, including 2-AG ether (noladin ether), O-arachidonoyl ethanolamine (virodhamine) and endogenous analogues of anandamide (eicosatrienoylethanolamide and docosatetraenoylethanolamide), were also identified (Freund et al. 2003, Pacher et al. 2006). Recent studies have also suggested that in addition to CB1R and CB2R, cannabinoid ligands can exert effects on other receptors, such as GPR55 and transient receptor potential vanilloid type 1 (Pacher et al. 2006). In the past decade, the endocannabinoid system has been implicated in a number of physiological functions in the central and peripheral nervous systems and in peripheral organs. Drugs acting on the endocannabinoid system have therapeutic potential in a number of pathologic conditions, including obesity and metabolic syndrome, mood and anxiety disorders, movement disorders, neuropathic pain, multiple sclerosis and spinal cord injury, as well as in atherosclerosis, myocardial infarction, stroke, hypertension, cancer, glaucoma and osteoporosis (Pacher et al. 2006, Table 1).
Affinities of the main cannabinoid ligands to CB1R
Rimonabant, an antagonist of CB1R, has already been introduced to the market to treat obesity and nicotine addiction (Bellocchio et al. 2006). Although the central side effects seriously limit the widespread use of CB1R antagonists (Steinberg & Cannon 2007), the therapeutical potential of drugs acting on CB1R is still very high (Kunos et al. 2008). The aim of this review is to discuss the intracellular mechanisms of the action of CB1R. After summarizing the available data about the G-protein activation and signal transduction of CB1R, this review also analyses recent data about dimerization or oligomerization of these receptors, and the tonic/basal activity of the receptor, which may contribute to its importance as a therapeutic target in metabolic diseases (Kunos et al. 2008).
Classical signal transduction pathways
Inhibition of cAMP production
Inhibition of adenylyl cyclase activity was the first characterized cannabinoid agonist-stimulated CB1R signal transduction pathway (Howlett & Fleming 1984, Howlett 1985, Howlett et al. 1986; Fig. 1A). The effect was blocked by pertussis toxin, indicating that it was mediated through Gi/o proteins. CB1R-mediated inhibition of adenylyl cyclase activity was reported in neural and peripheral tissues, as well as in cells overexpressing CB1R as reviewed previously (Howlett 2005). Coexpression of isoforms 1, 3, 5, 6 or 8 of adenylyl cyclase resulted in CB1R-mediated inhibition of cAMP accumulation, suggesting that the activated Gi/o proteins regulate these isoforms of the enzyme (Rhee et al. 1998). In recent years, numerous pharmacological studies have indicated that G-protein-coupled receptors (GPCRs) can adopt multiple conformations, leading to different signalling events. These different conformations can be stabilized by different ligands, causing ligand-biased signal transduction. In this concept, agonist and antagonist properties of a ligand can only be interpreted when a particular signal transduction pathway is defined, since the same ligand can behave as an agonist in one pathway, or antagonist or inverse agonist in another (Kenakin 2007). In the case of CB1R, similar biased agonism was also observed when activation of different Gi/o subtypes were measured (Houston & Howlett 1998, Glass & Northup 1999, Mukhopadhyay et al. 2002). In the membranes of Sf9 cells, the relative activation of Gi and Go was dependent on agonist (Glass & Northup 1999). In N18TG2 cells, WIN55 212-2 behaved as full agonist for Gi1, Gi2 and Gi3, while desacetyllevonantradol is agonist at Gi1 and Gi2 and inverse agonist at Gi3. Methanandamide, a more stable analogue of anandamide, acts as agonist at Gi3 and inverse agonist at Gi1 and Gi2. SR141716 was inverse agonist at all three G proteins tested by coimmunoprecipitation (Mukhopadhyay & Howlett 2005). In another study, it was demonstrated that WIN55 212-2 activated different Gi/o subtypes with different efficacies, suggesting that ligand-biased agonism is a more complicated issue as even a single ligand can activate different pathways depending on ligand concentration (Prather et al. 2000). Another complicating factor is that CB1R may have different affinity states, which may represent different conformations (Houston & Howlett 1998). In agreement with the concept of multiple receptor states, CB1R coupled with CP 55 940 or WIN 55 212-2 adopted different conformations as detected with plasmon-waveguide resonance spectroscopy (Georgieva et al. 2008). In addition, relative signalling efficacies of CB1R ligands may be also different in different brain regions, and the same ligand could activate Gi/o subtypes with different potencies in a number of brain regions (Sim et al. 1996, Breivogel et al. 1997, 2004, Breivogel & Childers 2000). These later results may be explained by different profiles of G proteins, or possibly with diverse interactions (e.g. dimerization) with other GPCRs (see below).
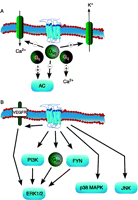
Main signal transduction pathways of CB1R activaton. G-protein activation and modulation of ion channels (A), and activation of different MAPK pathways (B).
Stimulation of cAMP production
It has been reported that in pertussis-pretreated cells, CB1R stimulation leads to adenylyl cyclase activation suggesting that in certain circumstances, CB1R can couple to Gs proteins (Glass & Felder 1997, Abadji et al. 1999, Calandra et al. 1999, Kearn et al. 2005; Fig. 1A). However, others could not immunoprecipitate G proteins, other than Gi/o, with CB1R (Mukhopadhyay et al. 2000). Coexpression of isoforms 2, 4 or 7 of adenylyl cyclase also resulted in CB1R-mediated stimulation of cAMP formation, but this effect was probably mediated by Gβγ dimers released from Gi proteins (Rhee et al. 1998).
Activation of Gq/11 proteins
Ca2+ signalling after CB1R stimulation has also been reported (Fig. 1A), however, the mechanism of this response is not clear. CB1R stimulation leads to an increase in Ca2+ levels in N18TG and NG108-15 cells, but not in C9 cells (Sugiura et al. 1996). It has also been reported that this response was pertussis toxin-sensitive in NG108-15 cells, suggesting the role of Gi/o proteins, which can regulate β2 isoform of phospholipase C (PLC) via Gβγ subunits, in this response (Sugiura et al. 1996, 1997). In contrast, in HEK cells expressing CB1R, only WIN55 212-2 caused Ca2+ signal generation, which occurred through activation of Gq proteins (Lauckner et al. 2005). Ca2+ signalling after stimulation of CB1R and CB2R has also been reported in insulinoma cells, and this response was Gq/PLC dependent and induced by arachidonoyl-chloro-ethanolamide and JWH133, a CB2R agonist (De Petrocellis et al. 2007). Anandamide also induced Ca2+ signal CB1R dependently. These cell-type-specific differences in the mechanism of CB1R-mediated Ca2+ signal generation may point to the different G-protein subunit composition of various cell types, or may be caused by different dimerization/interaction of CB1R with other receptors in these cells.
In contrast to CB1R and CB2R, GPR55, which has been suggested to be another cannabinoid receptor, seems to be coupled mainly to Ca2+ signalling (Baker et al. 2006, Ross 2009). It has been reported that GPR55 induces Ca2+ signal generation, but it is a lysophosphatidylinositol receptor (Oka et al. 2007). Another report showed, using GTPγS binding in HEK293 cells, that GPR55 was activated by CP 55 940, O-1602, THC, palmitoylethanolamide, anandamide, 2-AG, cannabinoidal (abnormal-CBD) and virodhamine, and was coupled to G13 protein, without activation of Ca2+ signal generation (Ryberg et al. 2007). In this report, WIN55 212-2 neither coupled nor activated GPR55, similar to findings of Johns et al. (2007). In contrast, in another report, GPR55 expressed in HEK293 cells, and its stimulation by THC, anandamide, methanandamide and JWH015, but not by 2-AG, palmitoylethanolamide, CP 55 940, virodhamine, and abnormal-CBD lead to Gq activation and Ca2+ signal generation (Lauckner et al. 2008). In endothelial cells, GPR55 was also coupled to Ca2+ signal, but it was dependent on CB1R activity (Waldeck-Weiermair et al. 2008). Since GPR55 and CB1R may have overlapping ligand specificity and, more importantly, both are inhibited by rimonabant (SR141716A), it has to be emphasized that calcium signals observed in natural tissues and inhibited by rimonabant (SR141716A) can also be mediated by GPR55. On the other hand, GPR55 may turn out not to be a cannabinoid receptor after all, since inverse agonists AM251 and rimonabant induce β-arrestin coupling to the receptor, suggesting that they activate it, while classical cannabinoids have no such or a weak effect (Yin et al. 2009).
Modulation of ion channels
Stimulation of CB1Rs leads to activation of G-protein-coupled inwardly-rectifying potassium channels (GIRKs) through pertussis-sensitive Gi/o proteins (Henry & Chavkin 1995, Mackie et al. 1995), and this activation can be induced by different CB1R agonists. In Xenopus laevis oocytes, GIRK1 and GIRK4 activation was reported, and this response was also detected in neuronal cells (Henry & Chavkin 1995, McAllister et al. 1999, Guo & Ikeda 2004, Azad et al. 2008).
CB1Rs can also influence the function of voltage-gated calcium channels. Inhibition of L-type calcium channels by cannabinoid stimulation was detected in cerebral vessels (Gebremedhin et al. 1999), in retinal bipolar cells (Straiker et al. 1999) and in neonatal rat solitary tract cells (Endoh 2006), whereas activation of these channels was reported in N18TG2 cells (Rubovitch et al. 2002). Inhibition of N-type calcium channels was detected in a number of experiments (Caulfield & Brown 1992, Mackie & Hille 1992, Felder et al. 1993, Mackie et al. 1993, Pan et al. 1996, Brown et al. 2004, Azad et al. 2008), and this effect may have a role in the presynaptic inhibition and retrograde signalling induced by cannabinoids (Freund et al. 2003, Howlett 2005). P/Q-type calcium channels are also negatively modulated by CB1R (Mackie et al. 1995, Hampson et al. 1998, Ho et al. 2000, Brown et al. 2004, Fisyunov et al. 2006).
β-Arrestin binding to CB1R
β-Arrestins were initially identified as proteins that play a role in the desensitization of GPCRs. Following receptor activation and subsequent phosphorylation by GPCR kinases (GRKs), β-arrestins bind to receptors and initialize their internalization. In addition to their role in regulation of GPCR internalization, β-arrestins can also serve as scaffolds for signalling complexes. The mechanism of β-arrestin binding to various GPCRs can be different. Relatively few data are available about the β-arrestin-binding properties of cannabinoid receptors. However, reports have indicated that β-arrestins play a role in desensitization of CB1Rs (Jin et al. 1999, Kouznetsova et al. 2002, Breivogel et al. 2008, Daigle et al. 2008b). It has been shown that expression of β-arrestin 2 and GRK3 in Xenopus oocyte accelerates desensitization of CB1Rs (Jin et al. 1999), and expression of dominant negative GRKs and β-arrestins interferes with CB1R desensitization in hippocampal neurons (Kouznetsova et al. 2002). Residues S426 and S430 were identified as responsible for β-arrestin-dependent desensitization of CB1Rs; however, mutations of these amino acids did not interfere with internalization of the receptor in AtT20 and HEK293 cells at least at early time points (Jin et al. 1999, Daigle et al. 2008a). Both wild-type and S426A/S430A mutant CB1Rs recruit β-arrestins in HEK293 cells following stimulation with CP 55 940 (Daigle et al. 2008a). In contrast, serine and threonine amino acids in the extreme carboxyl-terminal of CB1Rs are involved both in internalization and β-arrestin binding (Daigle et al. 2008b). Thus, it seems that different residues are responsible for β-arrestin-mediated desensitization and internalization; however, it cannot be ruled out that β-arrestin-independent CB1R internalization mechanisms are responsible for these different structural requirements. Receptor densities of CB1Rs in brain regions were not affected in β-arrestin knockout mice, and their desensitization was affected only when receptors were stimulated with THC (Breivogel et al. 2008), suggesting that control of receptor desensitization by β-arrestin may be dependent on agonist type. In conclusion, the data available today show that β-arrestins are involved in the desensitization of CB1R, although whether it has role in the internalization or G-protein-independent signalling remains to be elucidated.
Activation of mitogen-activated protein kinase pathways
Mitogen-activated protein kinase (MAPK) pathways are often activated after stimulation of GPCRs. GPCRs can regulate cell proliferation, cell differentiation, cell movement and cell death through MAPK. MAPK-signalling cascades are organized hierarchically, as MAPKs are activated by MAPK kinases (MAPKKs), which are activated by MAPKK kinases (MAPKKKs). MAPKKKs are activated by small GTPases or other protein kinases. MAPK cascades include pathways leading to activation ERK1/2, c-Jun N-terminal kinase (JNK), p38 MAPK or ERK5 proteins.
Stimulation of CB1R in vitro and in vivo leads to activation of ERK1/2 kinases in a variety of cell types (Howlett 2005). CB1R-mediated activation of ERK1/2 proteins can involve a number of mechanisms including activation of Gi/o proteins (Howlett 2005), phosphatidylinositol 3-kinase (PI3K; Galve-Roperh et al. 2002), transactivation of VEGF receptors (Korzh et al. 2008), through inhibition of adenylyl cyclase and protein kinase A (Davis et al. 2003) and the Src tyrosine kinase FYN (Derkinderen et al. 2003). Similar to G-protein activation, ERK1/2 stimulation also seems to be dependent on the particular cell type and different agonists can activate ERK1/2 through different pathways. CB1R stimulation is also followed by p38 MAPK and JNK activation (Liu et al. 2000, Rueda et al. 2000, Paradisi et al. 2008; Fig. 1B). It has been reported that CB1R activation can stimulate ERK1/2, p38 MAPK and JNK in endothelial cells (Liu et al. 2000).
In rat hippocampal slices, cannabinoids were able to activate p38 MAPK, but not JNK (Derkinderen et al. 2001). In cultured cortical neurons, JNK activation was observed when stimulated with THC (Downer et al. 2003), but no ERK1/2, p38 MAPK and JNK activation was observed after stimulation with another agonist, HU-210 (Molina-Holgado et al. 2005). These differences may reflect the dependence of these pathways on the maturation of neural cells (Downer et al. 2007). In Neuro2a cells, HU-210 activated only ERK1/2, but not JNK or p38 MAPK pathways (Graham et al. 2006), although in another study JNK activation through Src kinase was reported (He et al. 2005, 2006). In Chinese Hamster Ovary (CHO) cells, both JNK and p38 MAPK were activated after stimulation with THC. JNK activation was dependent on Gi/o proteins, PI3K and Ras, and involved platelet-derived growth factor (PDGF) receptor transactivation. On the other hand, p38 MAPK was not dependent on PDGF receptor activation (Rueda et al. 2000).
CB1R stimulation, in addition to direct modulation of MAPK pathways, may also regulate these pathways indirectly by modulating MAPK activation induced by other GPCRs (Ellis et al. 2006, Canals & Milligan 2008), or insulin and growth factor receptors (Bouaboula et al. 1997, Rajesh et al. 2008).
Other aspects of CB1R signalling
Homo- and heterodimerization of CB1R
Soon after identification of the β-adrenergic receptor, the physical interaction and cooperativity between receptor molecules were suggested (Limbird & Lefkowitz 1976). Since then, a large amount of data has accumulated suggesting that GPCRs function in dimers or in higher order oligomers (Bulenger et al. 2005, Waldhoer et al. 2005, Milligan et al. 2006, Milligan & Smith 2007, Szidonya et al. 2008). Strong evidences suggest that coexpression of two receptors leads to functional consequences; however, the exact nature of these interactions is difficult to determine using currently available techniques (Szidonya et al. 2008). It is usually accepted that there are direct interactions between GPCRs, however, the majority of data was obtained with biophysical, biochemical and structural methods that have been criticized by recent publications that challenge the concept. Despite these concerns, GPCR dimerization is a useful paradigm to explain the altered functional properties of these receptors in the presence of other GPCRs. Since dimerization can influence the signal transduction of GPCRs, it must be taken into consideration when the function of a GPCR is analyzed. In recent years, evidence has been presented that supports homo- or heterodimerization of CB1Rs. The first observation of the homodimerization of the CB1Rs was presented by Wager-Miller et al. (2002) using western blotting. These authors showed high molecular weight bands of CB1Rs, and suggested that these bands correspond to dimerized receptors (Wager-Miller et al. 2002). Another paper reported that CB1R and D2 dopamine receptors can form heterodimers using coimmunoprecipitation, and suggested that this dimerization tends to be dependent on the activity of CB1R (Kearn et al. 2005). However, activity-dependent localization of CB1R in different membrane compartments has also been reported (Sarnataro et al. 2006), which may affect the interpretation of the above-mentioned coimmunoprecipitation data. Although western blot and coimmunoprecipitation have been widely used for detection of GPCR dimers, these techniques may have several drawbacks including difficulties with solubilization of membranes, formation of GPCR aggregates, inappropriate selection of detergents, remaining membrane patches in supernatant and effects of the receptor glycosylation (Szidonya et al. 2008). Because of these and other difficulties, the data obtained with western blotting cannot be accepted as conclusive evidence for CB1R homodimerization, and data obtained with other methods are necessary to verify these findings. Using resonance energy transfer methods, heterodimerization of CB1R with D2 and A2A receptors (Carriba et al. 2008), opiate receptors (Rios et al. 2006) has been observed in transfected cells, and the functional role of these heterodimerizations has also been suggested (Hojo et al. 2008, Marcellino et al. 2008). Although bioluminescence resonance energy transfer (BRET) has also been widely used in dimerization studies, it needs careful experimental design and appropriate controls to conclude on specific interaction between two receptors (Marullo & Bouvier 2007). The pure BRET ratio cannot be interpreted as indicator of the proximity of two receptors and specificity of the interaction as it depends on many conditions including expression levels, expression ratios and the orientation of fluorophores. For example, the BRET ratio can be greatly influenced by simply changing the donor–acceptor ratio.
Another approach to study the interaction between GPCRs is detection of the functional changes caused by coexpression of receptors. When CB1R and μ-opioid receptors (μ-OR) were coexpressed, it has been shown that stimulation of one receptor can attenuate the signal transduction of the other receptor (Rios et al. 2006). Similarly, it has been reported that basal activity of CB1R also attenuates signalling of μ-OR (Canals & Milligan 2008). Although these interactions were suggested to be caused by dimerization, other alternative mechanisms may explain these findings, such as sequestration of G proteins (Bouaboula et al. 1997, Vasques & Lewis 1999, Nie & Lewis 2001, Chillakuri et al. 2007). Reciprocal inhibition between GABAB and CB1R has been reported in hippocampal membranes (Cinar et al. 2008). Another interesting interaction between Gi/o-coupled CB1R and D2-dopamine receptors has been reported (Kearn et al. 2005). Activation of both receptors decreased the forskolin-induced adenylyl cyclase activity; however, when the receptors were coexpressed, CB1R stimulation reversed the inhibition caused by D2 stimulation and led to an increase in cAMP levels. These data have been interpreted as a consequence of receptor heterodimerization; however, if we take into consideration that CB1R can couple to Gs proteins when Gi/o proteins are inhibited, the sequestration of Gi/o proteins cannot be ruled out as an alternative explanation for this finding. Another interesting functional interaction between CB1R and orexin-1 receptor was reported by Hilairet et al. (2003). They showed that CB1R coexpression increased the potency of orexin A to induce MAPK activation by orexin-1 receptor and this was blocked by CB1R antagonist. However, since orexin-1 is a Gq/11-coupled receptor, transactivation of CB1Rs by endocannabinoids and CB1R-mediated MAPK activation in this set-up cannot be ruled out. When the Gq-coupled AT1R was coexpressed with CB1R in CHO cells, AT1R stimulation with angiotensin II lead to a diacylglycerol lipase (DAGL)-mediated transactivation of CB1Rs (Turu et al. 2007). This effect was mediated by the formation of endocannabinoids in CHO cells, and similar CB1R transactivation was observed with all other tested Gq-coupled GPCRs (Turu et al. 2009). Since Gq/11 activation can lead to generation of 2-AG, the above-mentioned interaction between orexin-1 receptor and CB1R may also occur with this mechanism.
Perhaps the most convincing evidence for dimerization of GPCRs came from studies with GABAB receptors, because it has been shown convincingly that GABAB1 and GABAB2 work as a functional dimers in which GABAB1 binds the agonists and GABAB2 is responsible for efficient trafficking of the dimer and for coupling to the G proteins (Galvez et al. 2001). One of the consequences of dimerization could be coupled trafficking of GPCRs to and from the plasma membrane. Such coupled trafficking of CB1Rs has been recently demonstrated by Ellis et al. (2006), who showed that orexin-1 receptor distribution in the cells was markedly changed after coexpression of CB1R. It changed its localization from plasma membrane to intracellular vesicles, which is similar to the cellular distribution of CB1Rs. Both CB1R and orexin-1 antagonists externalized both receptors to the plasma membrane, and this observation is difficult to interpret differently than a direct interaction (dimerization or oligomerization) between receptors. Interestingly, on the other hand, the other previously suggested dimer pair of the CB1R, the μ-OR, failed to traffic together with CB1R in the same experimental setting (Ellis et al. 2006).
In conclusion, although the dimerization of GPCRs is an emerging important paradigm, it requires careful examination. In order to conclude the physiological relevance of the dimerization of GPCRs, coherent data obtained with biochemical, biophysical and pharmacological approaches should be assembled. The available data show clearly that coexpression of CB1R alters the function and signal transduction of other GPCRs, and vice versa. However, the data obtained with different methods are either lacking or contradictory, suggesting that firm interpretation of the available data as a consequence of dimerization or other type of interactions needs further investigations (Fig. 2).
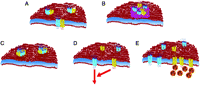
Possible mechanisms of interactions between GPCRs. Dimerization (A), clustering of GPCRs in specific membrane subdomains (B), higher order GPCR oligomers (C), interaction between GPCRs in signal transduction pathways (D) and sequestration of G-proteins (E).
Constitutive versus basal activity
Although a number of GPCRs show basal activity even in the absence of agonists, especially when overexpressed, the role of the constitutive activity in physiological conditions (when the receptor is not overexpressed) is known only in some cases (Seifert & Wenzel-Seifert 2002). The best example is the MC4 melanocortin receptor and its inverse agonist, the agouti-related peptide (Adan & Kas 2003), which has an extensively studied role in regulation of weight balance.
CB1R also shows a high degree of basal activity, both in expression systems and in native tissues and, as such, it is usually referred as a constitutively active receptor (Bouaboula et al. 1997, Rinaldi-Carmona et al. 1998, Pertwee 2005). However, in the case of receptors expressed in native tissues, the endogenous ligands released by the tissues may represent an alternative explanation to the high level of basal activity (Seifert & Wenzel-Seifert 2002). In many tissues, 2-AG, an endocannabinoid, is present in concentrations close to the Kd of CB1R (Sugiura et al. 2006). Moreover, DAGLs, the enzymes responsible for 2-AG generation, are present in most tissues, suggesting that endocannabinoid production could be a common property of different cell types (Bisogno et al. 2003). Despite these data, it has been widely accepted that the CB1R is constitutively active even in the absence of endocannabinoids, and there is some evidence that points to this conception, as reviewed recently (Pertwee 2005). Some other data suggest that the constitutive activity is not present in all CB1R expressing cells, or it can be inhibited by Ca2+ chelation (Katona et al. 1999, Hoffman & Lupica 2000, Wilson & Nicoll 2001, Chevaleyre & Castillo 2003, Savinainen et al. 2003, Breivogel et al. 2004, Hentges et al. 2005, Zhu & Lovinger 2005, Neu et al. 2006) or by inhibition of DAGL (Turu et al. 2007). There is also an observation that SR141716A, the CB1R-inverse agonist, can inhibit other receptors as well (Savinainen et al. 2003, Lauckner et al. 2008), and this can complicate the interpretation of its inverse agonist effects.
A pure neutral antagonist would help to decide at which degree is CB1R constitutively active. Indeed, there are some neutral CB1R antagonists in the literature (Pertwee 2005). Neutral antagonists have no effect on the constitutive CB1R activity, although they should inhibit it if endocannabinoids were the cause of basal activity. Three antagonists, NESS 0327, O-2654 and O-2050, do not show inverse agonist effects, although they do antagonize the agonists (Ruiu et al. 2003, Pertwee 2005, Canals & Milligan 2008). Similarly, cannabinol, another cannabis derivate, can antagonize the binding of CP 55 940, although it shows no inverse agonism (MacLennan et al. 1998). These observations argue against the role of endocannabinoids in the basal activity of CB1R.
Although these data would show that CB1R is constitutively active, the way how the neutral antagonist properties of these compounds were defined makes this issue a bit complicated. These compounds were named neutral antagonists because they do not change the basal activity of the CB1R in some experimental conditions. However, they can only be accepted as neutral antagonists if we accept that CB1R is constitutively active. If we would presume that the basal activity was caused by endocannabinoids present in the tissues, these compounds should be referred to as partial agonists, causing a similar degree of activation as endogenous cannabinoids. In concert with this, although the neutral antagonist effect of the compound O-2050 has not been described in publication, but is taken as such ligand (Martin et al. 2002, Pertwee 2005, Gardner & Mallet 2006, Canals & Milligan 2008); in the patent description, it has a similar partial agonist effect as THC, the first known cannabinoid agonist, when measured with [35S]-GTPγS binding on membranes (Martin et al. 2005). So these compounds are taken as neutral antagonists based on the observation that in certain experiments, they do not change the activity of CB1R. Since we assume that CB1R is constitutively active when we define these antagonists as neutral ones, results obtained with these ligands cannot be used as evidence of constitutive activity. To decide whether these compounds are really neutral antagonists, we need further experiments, maybe with mutated receptors that have altered basal activity.
In conclusion, although the CB1R is usually referred as a constitutively active receptor, the significant role of endocannabinoids in high basal activity cannot be ruled out (Fig. 3).
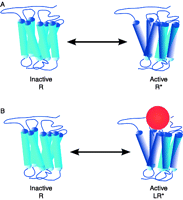
Two possible explanations of CB1R's basal activity: constitutive activity, when some receptors are present in active state even in the absence of any ligand (top); or endogenously produced agonists cause basal activity (bottom).
Declaration of interest
I declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by grants from the Hungarian Science Foundation (OTKA Grants NK-072661, M-045341), the National Development Agency, Hungary (TÁMOP 4.2.2-08/1/KM), and the Hungarian Ministry of Public Health.
- Revision received 19 June 2009
- Accepted 20 July 2009
- © 2010 Society for Endocrinology










