| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
|
|
||||||||
Review |
1 Departments of Medicine,
2 Molecular Physiology and Biophysics and
3 Program in Developmental Biology, Vanderbilt University Medical Center, 2220 Pierce Avenue 746 PRB, Nashville, Tennessee 37232, USA
(Requests for offprints should be addressed to M Gannon; Email: maureen.gannon{at}vanderbilt.edu)
| Abstract |
|---|
|
|
|---|
| Introduction |
|---|
|
|
|---|
| Pancreas development |
|---|
|
|
|---|
Pancreatic progenitors within the foregut epithelium are marked by the expression of pancreatic-duodenal homeobox 1 (Pdx1), which is induced at e8.5 in the foregut endoderm and is expressed throughout the pancreatic epithelium at e9.5 (Guz et al. 1995). Forkhead box A2 (FoxA2), previously called hepatic nuclear factor 3ß-(Hnf3ß ), can activate transcription of Pdx1, although there are many other upstream regulators of Pdx1 (Melloul et al. 2002). Pdx1 is required for growth, rather than formation, of the pancreatic buds, as evidenced by Pdx1–/– mice in which both pancreatic buds initially form but then arrest at a very early stage of development, resulting in an apancreatic phenotype at birth (Offield et al. 1996). The same phenotype has been observed in a human infant with a homozygous inactivating point mutation of PDX1 (Stoffers et al. 1997b). Pdx1 expression is downregulated in acinar and ductal cells beginning at approximately e13.0, but is maintained in differentiated endocrine cells and upregulated specifically in ß-cells (Guz et al. 1995). This expression pattern is maintained throughout life.
Pancreas transcription factor 1a (Ptf1a) is also expressed throughout the developing pancreas beginning at e9.5 and is required for the growth of the pancreatic buds (Krapp et al. 1998, Kawaguchi et al. 2002). Ptf1a is later downregulated in ductal and endocrine cells, but is maintained in acinar cells throughout life, where it induces expression of amylase and elastase. Ptf1a–/– mice form a rudimentary dorsal pancreas that fails to grow or produce differentiated acinar tissue (Krapp et al. 1998). Differentiated endocrine cells are present in these mice, although these cells are reduced in number and found scattered through the adjacent spleen.
| ß-Cell differentiation |
|---|
|
|
|---|
|
| In vivo ß-cell differentiation |
|---|
|
|
|---|
Differentiation of endocrine versus exocrine cells is accomplished by lateral inhibition within the ductal epithelium, mediated by the Notch signaling pathway (Apelqvist et al. 1999, Jensen et al. 2000). The pro-endocrine basic helix–loop–helix (bHLH) transcription factor neurogenin 3 (Ngn3), a downstream target of Hnf6 (Jacquemin et al. 2000), induces expression of Notch ligands, which bind to Notch receptors and activate the Notch pathway in adjacent cells (Heremans et al. 2002). Downstream targets of the Notch pathway (e.g. Hairy/enhancer-of-split 1, Hes1) repress Ngn3 expression, which inhibits endocrine differentiation. Additionally, Hes1 represses the cell cycle inhibitor p57, which maintains a proliferative pool of pancreatic progenitor cells within the embryonic ductal epithelium (Georgia et al. 2006). Therefore, cells in which the Notch signaling pathway is activated, maintain their proliferative capacity, while cells in which Notch signaling is not activated, express Ngn3, exit the cell cycle, and differentiate into endocrine cells. Genetic mouse models of impaired Notch signaling (Dll1–/–, Rbp-J –/–, Hes1–/–) exhibit increased endocrine cell differentiation at the expense of the pancreatic progenitor population (Apelqvist et al. 1999, Jensen et al. 2000).
Ngn3 is required for endocrine cell differentiation, as evidenced by Ngn3–/–mice, which lack all pancreatic endocrine cell types and die postnatally due to severe diabetes (Gradwohl et al. 2000). Additionally, lineage tracing analysis reveals that all endocrine cells arise from Ngn3+cells (Gu et al. 2002). Ngn3 induces expression of essential ß-cell transcription factors including neurogenic differentiation 1 (Neurod1), also known as Beta2 (Huang et al. 2000), and paired box gene 4 (Pax4; Smith et al. 2003), but is not itself expressed in hormone-producing cells. The bHLH transcription factor NeuroD1 induces expression of several endocrine genes, including insulin, and its expression is maintained in mature endocrine cells (Naya et al. 1995). NeuroD1–/– mice are diabetic due to severely reduced numbers of all endocrine cell types (Naya et al. 1997), and humans with heterozygous mutations in NEUROD1 suffer from a type of diabetes referred to as maturity-onset diabetes of the young type 6 (MODY6; Kristinsson et al. 2001). In contrast, ectopic expression of Ngn3 or NeuroD1 in the pancreatic epithelium results in premature and expansive differentiation of endocrine cells, primarily glucagon-producing -cells, at the expense of the pancreatic progenitor pool, resulting in a hypoplastic pancreas (Apelqvist et al. 1999, Schwitzgebel et al. 2000).
Endocrine cells produced by the second wave of differentiation can first be identified at e13.5, forming endocrine cords adjacent to ducts (Pictet et al. 1972). These cells delaminate from the ductal epithelium, differentiate, proliferate, and then cluster to form islets, which contain ß-cells, -cells, somatostatin-producing -cells, pancreatic polypeptide-producing PP-cells, and ghrelin-producing -cells. The regulation of differentiation down each of these endocrine lineages from a Ngn3+ cell is complex and still not completely understood. Lineage tracing analysis has revealed that the ß- and -cell lineages diverge early, while ß-cells and PP-cells may differentiate from the same lineage (Herrera 2000). Additionally, ß- and -cells share a requirement for Pax4, as exhibited by Pax4–/– mice, which have increased numbers of -cells at the expense of ß- and -cells (Sosa-Pineda et al. 1997). Pax4 is expressed in early insulin+, but not glucagon+, cells and is later restricted to mature ß-cells, but Pax4 alone is not sufficient to drive Ngn3+ cells towards either a ß- or -cell fate (Grapin-Botton et al. 2001). A putative antagonist of Pax4 is aristaless-related homeobox (Arx), whose expression pattern and loss-of-function phenotype directly contrast with that of Pax4 (Collombat et al. 2003). Arx is expressed in pancreatic progenitor cells beginning at e9.5 and is later restricted to mature - and -cells. Arx–/– mice exhibit increased numbers of ß- and -cells at the expense of -cells. Furthermore, Arx is upregulated in Pax4–/– mice, while Pax4 is upregulated in Arx–/– mice. Thus, Pax4 and Arx function in the differential specification of endocrine cell types.
Early broad expression of Pdx1 induces expression of the NK homeodomain transcription factors Nkx2.2 and Nkx6.1, but while Nkx2.2 expression becomes restricted to -, ß-, and PP-cells (Sussel et al. 1998), Nkx6.1 expression is tightly restricted to ß-cells (Sander et al. 2000). Nkx2.2–/– mice reveal that Nkx2.2 is absolutely required for ß-cell differentiation and plays a lesser role in differentiation of - and PP-cells (Sussel et al. 1998), while Nkx6.1–/– mice have impaired but not complete loss of ß-cell differentiation (Sander et al. 2000). Several pieces of evidence suggest that Nkx2.2 is upstream of Nkx6.1: initiation of expression of Nkx2.2 precedes that of Nkx6.1 (e9.5 vs e10.5 respectively); all Nkx6.1+ cells also express Nkx2.2, while not all Nkx2.2+ cells express Nkx6.1; Nkx2.2–/– mice lack expression of Nkx6.1; and Nkx2.2–/–; Nkx6.1–/–mice exhibit a similar phenotype to Nkx2.2–/– mice.
In addition to its early role in pancreas development, Pdx1 plays a role in the terminal differentiation of ß-cells by inducing expression of insulin, glucose transporter 2 (Glut2), glucokinase, and islet amyloid polypeptide (Iapp; Edlund 2001) and is necessary for maintaining mature ß-cell function. The basic-leucine zipper (bZIP) transcription factor MafA has also recently been identified as an important regulator of ß-cell function and a marker of mature ß-cells (Zhang et al. 2005a). MafA–/– mice undergo normal pancreatic development but develop diabetes postnatally, associated with progressively impaired insulin secretion, abnormal islet morphology, and reduced expression of insulin, Pdx1, NeuroD1, and Glut2. Additionally, insulin has been shown to be a direct transcriptional target of MafA (Kataoka et al. 2002, Olbrot et al. 2002, Matsuoka et al. 2004).
| In vitro ß-cell differentiation |
|---|
|
|
|---|
|
| Establishing an organism’s ß-cell mass |
|---|
|
|
|---|
|
ß-Cell apoptosis occurs at very low rates during embryogenesis, but there is evidence that a transient burst of ß-cell apoptosis occurs at weaning, which may be associated with islet remodeling and/or changes in ß-cell maturation (Scaglia et al. 1997). During adulthood, ß-cell apoptosis also normally occurs at very low rates. Average ß-cell size is fairly stable during the postnatal period (Scaglia et al. 1997), but it increases with age during later adulthood (Montanya et al. 2000).
Proper development of an organism’s ß-cell mass requires appropriate nutrition during embryogenesis. Poor maternal nutrition results in intrauterine growth retardation (IUGR), low birth weight, and under-developed ß-cell mass in newborn rats, which predisposes them to glucose intolerance and diabetes later in life (Breant et al. 2006). When IUGR is caused by total caloric restriction, the reduction in ß-cell mass is due to reduced ß-cell differentiation, with decreased expression of Pdx1, Pax6, and Nkx6.1, rather than due to reduced ß-cell proliferation. These changes are associated with increased levels of glucocorticoids, which can independently reduce fetal ß-cell mass when exogenously administered to the mother. IUGR caused by protein restriction also results in underdeveloped ß-cell mass in newborn rats, but this is due to reduced ß-cell proliferation and increased ß-cell apoptosis, associated with reduced levels of insulin-like growth factors (IGFs; Reusens & Remacle 2006). In contrast, maternal obesity and/or diabetes results in newborn macrosomia (large birth weight) and increased ß-cell mass associated with increased ß-cell proliferation, likely in response to maternal hyperglycemia. These offspring are predisposed to obesity, insulin resistance, and diabetes, associated with early ß-cell exhaustion.
| Maintaining an organism’s ß-cell mass |
|---|
|
|
|---|
The ability of an organism to maintain its ß-cell mass during adulthood is paramount to maintaining glucose homeostasis and preventing diabetes. Table 1![]() summarizes the mouse models in which molecular regulators of postnatal ß-cell mass are perturbed. Several genetic mouse models of cell cycle dysregulation impair postnatal ß-cell proliferation, and cause a progressive decline in ß-cell mass, associated with a progressive glucose intolerant and diabetic phenotype. For example, global inactivation of cyclin-dependent kinase 4 in mice (Cdk4–/–) specifically affects endocrine cells within the pancreas, causing diabetes by 2 months of age, and within the pituitary, causing reduced body size and infertility (Rane et al. 1999). The diabetes observed in these mice is associated with severely reduced ß-cell mass, although ß-cell mass is comparable with that of wild-type mice at postnatal days (P) 1 and 2. Progression from G1 to S phase in the cell cycle requires phosphorylation of the retinoblastoma protein (Rb) by Cdk4/6 complexed with a D cyclin, which releases E2F allowing it to activate transcription of necessary cell cycle target genes. Thus, Cdk6 function is redundant with Cdk4; however, ß-cells do not express detectable levels of Cdk6 (Martin et al. 2003), making them uniquely susceptible to cell cycle perturbations caused by loss of Cdk4. Similarly, global deletion of cyclin D2 (CyclinD2–/–), the predominant D cyclin expressed in ß-cells, fails to stimulate adequate compensatory up-regulation of cyclin D1 or D3 within islets and drastically impairs postnatal ß-cell proliferation (Georgia & Bhushan 2004, Kushner et al. 2005a). CyclinD2–/– mice exhibit normal ß-cell mass at e17.5 but significantly reduced ß-cell mass postnatally, which causes progressive glucose intolerance and diabetes.
summarizes the mouse models in which molecular regulators of postnatal ß-cell mass are perturbed. Several genetic mouse models of cell cycle dysregulation impair postnatal ß-cell proliferation, and cause a progressive decline in ß-cell mass, associated with a progressive glucose intolerant and diabetic phenotype. For example, global inactivation of cyclin-dependent kinase 4 in mice (Cdk4–/–) specifically affects endocrine cells within the pancreas, causing diabetes by 2 months of age, and within the pituitary, causing reduced body size and infertility (Rane et al. 1999). The diabetes observed in these mice is associated with severely reduced ß-cell mass, although ß-cell mass is comparable with that of wild-type mice at postnatal days (P) 1 and 2. Progression from G1 to S phase in the cell cycle requires phosphorylation of the retinoblastoma protein (Rb) by Cdk4/6 complexed with a D cyclin, which releases E2F allowing it to activate transcription of necessary cell cycle target genes. Thus, Cdk6 function is redundant with Cdk4; however, ß-cells do not express detectable levels of Cdk6 (Martin et al. 2003), making them uniquely susceptible to cell cycle perturbations caused by loss of Cdk4. Similarly, global deletion of cyclin D2 (CyclinD2–/–), the predominant D cyclin expressed in ß-cells, fails to stimulate adequate compensatory up-regulation of cyclin D1 or D3 within islets and drastically impairs postnatal ß-cell proliferation (Georgia & Bhushan 2004, Kushner et al. 2005a). CyclinD2–/– mice exhibit normal ß-cell mass at e17.5 but significantly reduced ß-cell mass postnatally, which causes progressive glucose intolerance and diabetes.
|
Our laboratory has recently found that FoxM1 also plays an important role in ß-cell proliferation (Zhang et al. 2006). FoxM1 is known to regulate expression levels of several cell cycle proteins: cyclin B, which complexes with Cdk1 to promote G2 to M phase progression; Cdc25A and B phosphatases, which dephosphorylate and activate Cdks; and S-phase kinase-associated protein 2 (Skp2) and Cdk subunit 1 (Cks1), which together form the Skp1-Cullin1-F-box protein (SCF) ubiquitin ligase complex that targets p27Kip1 and p21Cip1 for proteasomal degradation. Pancreas-specific deletion of Foxm1 using a Cre-lox strategy (Foxm1flox/flox; Pdx14.3-Cre) in mice results in postnatal defects in ß-cell proliferation, which contribute to a postnatal deficiency in ß-cell mass and a progressive glucose intolerant and diabetic phenotype. However, ß-cell mass and islet morphology are normal at P1, despite inactivation of Foxm1 early in embryogenesis. Increased nuclear p27Kip1, an indirect target of FoxM1, is associated with impaired ß-cell proliferation in these mice. Thus, FoxM1 is critical for maintaining ß-cell mass during adulthood by properly coordinating cell cycle progression.
Another regulator of cell cycle genes is the transcriptional co-activator menin, encoded by Men1, mutation of which results in multiple endocrine neoplasia type 1 (MEN1) in humans (Crabtree et al. 2003). This syndrome is characterized by hyperplasia of endocrine cell types primarily within the parathyroids, anterior pituitary, and pancreas, resulting in tumor formation, most commonly insulinomas composed of ß-cells. Men1+/– mice display a similar phenotype. ß-Cell-specific deletion of Men1 (Men1flox/flox; Rip-Cre) also results in ß-cell hyperplasia, insulinomas, hyperinsulinemia, and hypoglycemia. Significantly increased rates of ß-cell proliferation are observed, and this is associated with reduced levels of p18INK4c and p27Kip1, both of which are direct targets of menin-mediated histone methylation (Karnik et al. 2005). Furthermore, a MEN1-like phenotype is observed in p18INK4c–/–; p27Kip1–/– mice (Franklin et al. 2000). Other mouse models of impaired and enhanced ß-cell proliferation have been reviewed by Cozar-Castellano et al. (2006b) and Heit et al. (2006b).
Just as impaired ß-cell proliferation causes a net loss of ß-cells, increased ß-cell death can have the same effect. Inherent defects that make ß-cells more susceptible to apoptosis, for example, result in a negative balance of ß-cell turnover, as observed in Pdx1+/– mice, which exhibit normal ß-cell mass at 3 months of age, but are unable to appropriately increase their ß-cell mass as they age (Johnson et al. 2003). Haploinsufficiency of Pdx1 makes ß-cells more susceptible to undergoing apoptosis, associated with reduced expression levels of the antiapoptotic genes BclXL and Bcl-2. Increased ß-cell apoptosis results in insufficient ß-cell mass and progressive glucose intolerance. A more dramatic phenotype is observed in mice with ß-cell-specific deletion of Pdx1 (Pdx1flox/flox; Rip-Cre; Ahlgren et al. 1998). These mice suffer from worsening glucose intolerance due to both progressive loss of ß-cell mass and impaired ß-cell function. Mutations of PDX1 in humans result in similar phenotypes; a dominant-negative mutation of PDX1 causes MODY4 (Stoffers et al. 1997a), while a heterozygous inactivating mutation of PDX1 predisposes to late-onset type II diabetes (Macfarlane et al. 1999). Thus, Pdx1 is a critical regulator of ß-cell survival and maintenance of ß-cell mass and function during adulthood.
| Dynamic changes in an organism’s ß-cell mass |
|---|
|
|
|---|
During pregnancy, rats exhibit a greater than 50% increase in ß-cell mass, which is accomplished primarily through an approximate threefold increase in ß-cell proliferation (Scaglia et al. 1995). The chief stimuli of ß-cell proliferation during pregnancy are placental lactogens (PLs), although prolactin (Prl) and growth hormone (GH) also have similar effects on ß-cells and are also elevated during pregnancy. After delivery, ß-cell mass returns to normal levels within 10 days through increased ß-cell apoptosis, decreased ß-cell proliferation, and ß-cell atrophy.
To determine the direct role of PLs on ß-cell mass in non-pregnant animals, transgenic mice expressing PL1 within their ß-cells (Rip-Pl1) were developed (Vasavada et al. 2000, Cozar-Castellano et al. 2006a). These mice exhibit hypoglycemia and improved glucose clearance due to hyperinsulinemia, which is associated with a doubling of ß-cell mass. This expansion of ß-cell mass is attributed to a twofold increase in ß-cell proliferation and a 20% increase in ß-cell size. Similar results are observed in transgenic mice expressing HGF within their ß-cells (Rip-Hgf; Garcia-Ocaña et al. 2000, 2001, Cozar-Castellano et al. 2006a). These mice also exhibit a doubling of ß-cell mass, but the increase in ß-cell proliferation is not as significant as that in Rip-Pl1 mice. Interestingly, islet number is significantly increased in Rip-Hgf mice but not in Rip-Pl1 mice versus wild-type mice, suggesting that HGF may stimulate neogenesis.
Diet-induced obesity results in insulin resistance and ß-cell mass expansion in humans and mice. The C57Bl/6 mouse strain is notoriously susceptible to these effects, exhibiting a 2.2-fold increase in ß-cell mass and proliferation after 4 months on a high-fat diet versus a control diet (Sone & Kagawa 2005). However, these mice eventually become diabetic and lose their ß-cell mass due to increased ß-cell apoptosis and reduced ß-cell proliferation.
In genetic models of obesity and insulin resistance, there is also a compensatory expansion of ß-cell mass. For example, db/db mice, which lack a functional leptin receptor, exhibit a twofold increase in ß-cell mass by 8 weeks of age (Wang & Brubaker 2002). This timepoint correlates with the onset of diabetes, which progresses from glucose intolerance that is first observed between 4 and 6 weeks of age. A similar rat model, the Zucker diabetic fatty (ZDF) rat (fa/fa), also has a homozygous mutation in the gene encoding the leptin receptor. ZDF rats exhibit increased ß-cell mass and increased ß-cell proliferation prior to the onset of diabetes, but increased ß-cell apoptosis prevents them from adequately expanding their ß-cell mass after the onset of diabetes, despite continued high rates of ß-cell proliferation (Pick et al. 1998). This phenotype contrasts with what is observed in non-diabetic Zucker fatty (ZF) rats, which possess the same mutation as ZDF rats and also become obese and insulin resistant but do not develop diabetes due to sufficient ß-cell mass expansion through increased ß-cell proliferation, neogenesis, and hypertrophy (Pick et al. 1998).
Another model of insufficient ß-cell mass expansion is the insulin receptor substrate two null mouse (Irs2–/–; Kubota et al. 2004). Global inactivation of Irs2 results in severe insulin resistance, both centrally in the brain causing obesity, and peripherally, for which ß-cell mass expansion should be able to compensate. However, because ß-cells require Irs2 for proper proliferation and function, Irs2–/–mice are unable to expand their ß-cell mass, and they develop diabetes by 10 weeks of age. This phenotype is not observed in Irs1–/–mice, despite the fact that these mice exhibit similar insulin resistance, because Irs1 is not required for ß-cell mass expansion. Deletion of Irs2 specifically within ß-cells and the hypothalamus (Irs2flox/flox; Rip-Cre) causes central insulin resistance and obesity, with glucose intolerance developing at 8 weeks of age but without progression to diabetes, likely due to the lack of peripheral insulin resistance (Kubota et al. 2004). These mice also exhibit reduced ß-cell proliferation and ß-cell mass at 8 weeks of age, although these impairments are not observed before the onset of insulin resistance (between 4 and 8 weeks). These experiments provide additional evidence that Irs2 is required for ß-cell mass expansion in response to insulin resistance. Furthermore, overexpression of Irs2 in ß-cells (Rip-Irs2) is sufficient to prevent ß-cell failure in diet-induced obesity and streptozotocin-induced diabetic models (Hennige et al. 2003).
Several downstream effectors of the insulin signaling pathway have been shown to play a role in ß-cell mass expansion in models of insulin resistance. For example, haploinsufficiency of FoxO1 (Foxo1+/–) restores ß-cell mass and proliferation to nearly normal levels in Irs2–/– mice, possibly due to reduced FoxO1-mediated repression of Pdx1 (Kitamura et al. 2002). In contrast, expression of constitutively nuclear FoxO1 prevents ß-cell mass expansion in two other models of insulin resistance (Okamoto et al. 2006).
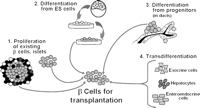
| Therapeutic implications |
|---|
|
|
|---|
| Acknowledgements |
|---|
| References |
|---|
|
|
|---|
Ahlgren U, Jonsson J, Jonsson L, Simu K & Edlund H 1998 Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes and Development 12 1763–1768.
Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U & Edlund H 1999 Notch signalling controls pancreatic cell differentiation. Nature 400 877–881.[CrossRef][Medline]
Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM & Permutt MA 2001 Islet beta cell expression of constitutively active Akt1/PKBalpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. Journal of Clinical Investigation 108 1631–1638.[CrossRef][Web of Science][Medline]
Bernal-Mizrachi E, Fatrai S, Johnson JD, Ohsugi M, Otani K, Han Z, Polonsky KS & Permutt MA 2004 Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. Journal of Clinical Investigation 114 928–936.[CrossRef][Web of Science][Medline]
Bernard-Kargar C & Ktorza A 2001 Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes 50 S30–S35.
Bhushan A, Itoh N, Kato S, Thiery JP, Czernichow P, Bellusci S & Scharfmann R 2001 Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development 128 5109–5117.
Blyszczuk P, Czyz J, Kania G, Wagner M, Roll U, St-Onge L & Wobus AM 2003 Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. PNAS 100 998–1003.
Bonner-Weir S & Weir GC 2005 New sources of pancreatic beta-cells. Nature Biotechnology 23 857–861.[CrossRef][Web of Science][Medline]
Boushey RP, Abadir A, Flamez D, Baggio LL, Li Y, Berger V, Marshall BA, Finegood D, Wang TC, Schuit F et al. 2003 Hypoglycemia, defective islet glucagon secretion, but normal islet mass in mice with a disruption of the gastrin gene. Gastroenterology 125 1164–1174.[CrossRef][Medline]
Breant B, Gesina E & Blondeau B 2006 Nutrition, glucocorticoids and pancreas development. Hormone Research 65 98–104.[CrossRef][Medline]
Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G & Gruss P 2003 Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes and Development 17 2591–2603.
Cozar-Castellano I, Weinstock M, Haught M, Velazquez-Garcia S, Sipula D & Stewart AF 2006a Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes 55 70–77.
Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocaña A, Vasavada R & Stewart AF 2006b Molecular control of cell cycle progression in the pancreatic beta-cell. Endocrine Reviews 27 356–370.
Crabtree JS, Scacheri PC, Ward JM, McNally SR, Swain GP, Montagna C, Hager JH, Hanahan D, Edlund H, Magnuson MA et al. 2003 Of mice and MEN1: insulinomas in a conditional mouse knockout. Molecular and Cellular Biology 23 6075–6085.
D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK & Baetge EE 2006 Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nature Biotechnology 24 1392–1401.[CrossRef][Web of Science][Medline]
Devedjian J-C, George M, Casellas A, Pujol A, Visa J, Pelegrin M, Gros L & Bosch F 2000 Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. Journal of Clinical Investigation 105 731–740.[Web of Science][Medline]
Dor Y, Brown J, Martinez OI & Melton DA 2004 Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429 41–46.[CrossRef][Medline]
Edlund H 2001 Developmental biology of the pancreas. Diabetes 50 S5–S9.
Fajas L, Annicotte J-S, Miard S, Sarruf D, Watanabe M & Auwerx J 2004 Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1–/– mice. Journal of Clinical Investigation 113 1288–1295.[CrossRef][Web of Science][Medline]
Fatrai S, Elghazi L, Balcazar N, Cras-Meneur C, Krits I, Kiyokawa H & Bernal-Mizrachi E 2006 Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes 55 318–325.
Finegood DT, Scaglia L & Bonner-Weir S 1995 Dynamics of beta-cell mass in the growing rat pancreas: estimation with a simple mathematical model. Diabetes 44 249–256.[Abstract]
Franklin DS, Godfrey VL, O’Brien DA, Deng C & Xiong Y 2000 Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Molecular and Cellular Biology 20 6147–6158.
Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, Kendall W, Oden J, Bridges S, Binart N et al. 2002 Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 143 1378–1385.
Fujikawa T, Oh SH, Pi L, Hatch HM, Shupe T & Petersen BE 2005 Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. American Journal of Pathology 166 1781–1791.
Gannon G, Mandriota SJ, Cui L, Baetens D, Pepper MS & Christofori G 2002 Overexpression of vascular endothelial growth factor-A165 enhances tumor angiogenesis but not metastasis during beta-cell carcinogenesis. Cancer Research 62 603–608.
Garcia-Ocaña A, Takane KK, Syed MA, Philbrick WM, Vasavada RC & Stewart AF 2000 Hepatocyte growth factor overexpression in the islet of transgenic mice increases beta cell proliferation, enhances islet mass, and induces mild hypoglycemia. Journal of Biological Chemistry 275 1226–1232.
Garcia-Ocaña A, Vasavada RC, Cebrian A, Reddy V, Takane KK, Lopez-Talavera J-C & Stewart AF 2001 Transgenic overexpression of hepatocyte growth factor in the beta-cell markedly improves islet function and islet transplant outcomes in mice. Diabetes 50 2752–2762.
Gasa R, Mrejen C, Leachman N, Otten M, Barnes M, Wang J, Chakrabarti S, Mirmira R & German M 2004 Proendocrine genes coordinate the pancreatic islet differentiation program in vitro. PNAS 101 13245–13250.
Georgia S & Bhushan A 2004 Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. Journal of Clinical Investigation 114 963–968.[CrossRef][Web of Science][Medline]
Georgia S & Bhushan A 2006 p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes 55 2950–2956.
George M, Ayuso E, Casellas A, Costa C, Devedjian JC & Bosch F 2002 Beta cell expression of IGF-I leads to recovery from type 1 diabetes. Journal of Clinical Investigation 109 1153–1163.[CrossRef][Web of Science][Medline]
Georgia S, Soliz R, Li M, Zhang P & Bhushan A 2006 p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Developmental Biology 298 22–31.[CrossRef][Web of Science][Medline]
Gradwohl G, Dierich A, LeMeur M & Guillemot F 2000 Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. PNAS 97 1607–1611.
Grapin-Botton A, Majithia AR & Melton DA 2001 Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes and Development 15 444–454.
Gu G, Dubauskaite J & Melton DA 2002 Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129 2447–2457.[Web of Science][Medline]
Guz Y, Montminy M, Stein R, Leonard J, Gamer L, Wright C & Teitelman G 1995 Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development 121 11–18.[Abstract]
Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD & Ron D 2001 Diabetes mellitus and exocrine pancreatic dysfunction in Perk–/–mice reveals a role for translational control in secretory cell survival. Molecular Cell 7 1153–1163.[CrossRef][Web of Science][Medline]
Harrison KA, Thaler J, Pfaff SL, Gu H & Kehrl JH 1999 Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nature Genetics 23 71–75.[Web of Science][Medline]
Hart AW, Baeza N, Apelqvist A & Edlund H 2000 Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature 408 864–868.[CrossRef][Medline]
Hashimoto N, Kido Y, Uchida T, Asahara S-I, Shigeyama Y, Matsuda T, Takeda A, Tsuchihashi D, Nishizawa A, Ogawa W et al. 2006 Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nature Genetics 38 589–593.[CrossRef][Web of Science][Medline]
Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR & Kim SK 2006a Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 443 345–349.[CrossRef][Medline]
Heit JJ, Karnik SK & Kim SK 2006b Intrinsic regulators of pancreatic beta-cell proliferation. Annual Review of Cell and Developmental Biology 22 311–338.[CrossRef][Web of Science][Medline]
Hennige AM, Burks DJ, Ozcan U, Kulkarni RN, Ye J, Park S, Schubert M, Fisher TL, Dow MA, Leshan R et al. 2003 Upregulation of insulin receptor substrate-2 in pancreatic beta cells prevents diabetes. Journal of Clinical Investigation 112 1521–1532.[CrossRef][Web of Science][Medline]
Heremans Y, Van De Casteele M, in’t Veld P, Gradwohl G, Serup P, Madsen O, Pipeleers D & Heimberg H 2002 Recapitulation of embryonic neuroendocrine differentiation in adult human pancreatic duct cells expressing neurogenin 3. Journal of Cell Biology 159 303–312.
Herrera PL 2000 Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127 2317–2322.[Abstract]
Huang H-P, Liu M, El-Hodiri HM, Chu K, Jamrich M & Tsai M-J 2000 Regulation of the pancreatic islet-specific gene BETA2 (neuroD) by neurogenin 3. Molecular and Cellular Biology 20 3292–3307.
Hussain MA, Porras DL, Rowe MH, West JR, Song WJ, Schreiber WE & Wondisford FE 2006 Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Molecular and Cellular Biology 26 7747–7759.
Iglesias A, Murga M, Laresgoiti U, Skoudy A, Bernales I, Fullaondo A, Moreno B, Lloreta J, Field SJ, Real FX et al. 2004 Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double-mutant mice. Journal of Clinical Investigation 113 1398–1407.[CrossRef][Web of Science][Medline]
Inada A, Hamamoto Y, Tsuura Y, Miyazaki J-I, Toyokuni S, Ihara Y, Nagai K, Yamada Y, Bonner-Weir S & Seino Y 2004 Overexpression of inducible cyclic AMP early repressor inhibits transactivation of genes and cell proliferation in pancreatic beta cells. Molecular and Cellular Biology 24 2831–2841.
Inoue M, Hager JH, Ferrara N, Gerber H-P & Hanahan D 2002 VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 1 193–202.[CrossRef][Web of Science][Medline]
Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D et al. 2000 Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Molecular and Cellular Biology 20 4445–4454.
Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P & Madsen OD 2000 Control of endodermal endocrine development by Hes-1. Nature Genetics 24 36–44.[CrossRef][Web of Science][Medline]
Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M & Montminy M 2003 cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes and Development 17 1575–1580.
Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, Misler S, Edlund H & Polonsky KS 2003 Increased islet apoptosis in Pdx1+/– mice. Journal of Clinical Investigation 111 1147–1160.[CrossRef][Web of Science][Medline]
Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M & Kim SK 2005 Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. PNAS 102 14659–14664.
Kassem SA, Ariel I, Thornton PS, Hussain K, Smith V, Lindley KJ, Aynsley-Green A & Glaser B 2001 p57KIP2 expression in normal islet cells and in hyperinsulinism of infancy. Diabetes 50 2763–2769.
Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M & Handa H 2002 MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. Journal of Biological Chemistry 277 49903–49910.
Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ & Wright CV 2002 The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nature Genetics 32 128–134.[CrossRef][Web of Science][Medline]
Kim SK & MacDonald RJ 2002 Signaling and transcriptional control of pancreatic organogenesis. Current Opinion in Genetics and Development 12 540–547.[CrossRef][Web of Science][Medline]
Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH III, Wright CVE, White MF, Arden KC & Accili D 2002 The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. Journal of Clinical Investigation 110 1839–1847.[CrossRef][Web of Science][Medline]
Krakowski M, Kritzik M, Jones E, Krahl T, Lee J, Arnush M, Gu D, Mroczkowski B & Sarvetnick N 1999 Transgenic expression of epidermal growth factor and keratinocyte growth factor in beta-cells results in substantial morphological changes. Journal of Endocrinology 162 167–175.[Abstract]
Krapp A, Knofler M, Ledermann B, Burki K, Berney C, Zoerkler N, Hagenbuchle O & Wellauer PK 1998 The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes and Development 12 3752–3763.
Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S & Sharpless NE 2006 p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443 453–457.[CrossRef][Medline]
Kristinsson SY, Thorolfsdottir ET, Talseth B, Steingrimsson E, Thorsson AB, Helgason T, Hreidarsson AB & Arngrimsson R 2001 MODY in Iceland is associated with mutations in HNF-1a and a novel mutation in NeuroD1. Diabetologia 44 2098–2103.[CrossRef][Web of Science][Medline]
Ku HT, Zhang N, Kubo A, O’Connor R, Mao M, Keller G & Bromberg JS 2004 Committing embryonic stem cells to early endocrine pancreas in vitro. Stem Cells 22 1205–1217.[CrossRef][Web of Science][Medline]
Kubota N, Tobe K, Terauchi Y, Eto K, Yamauchi T, Suzuki R, Tsubamoto Y, Komeda K, Nakano R, Miki H et al. 2000 Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 49 1880–1889.[Abstract]
Kubota N, Terauchi Y, Tobe K, Yano W, Suzuki R, Ueki K, Takamoto I, Satoh H, Maki T, Kubota T et al. 2004 Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. Journal of Clinical Investigation 114 917–927.[CrossRef][Web of Science][Medline]
Kulkarni RN, Holzenberger M, Shih DQ, Ozcan U, Stoffel M, Magnuson MA & Kahn CR 2002 Beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nature Genetics 31 111–115.[Web of Science][Medline]
Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P & White MF 2005a Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Molecular and Cellular Biology 25 3752–3762.
Kushner JA, Simpson L, Wartschow LM, Guo S, Rankin MM, Parsons R & White MF 2005b Phosphatase and tensin homolog regulation of islet growth and glucose homeostasis. Journal of Biological Chemistry 280 39388–39393.
Li H, Arber S, Jessell TM & Edlund H 1999 Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nature Genetics 23 67–70.[Web of Science][Medline]
Ling Z, Wu D, Zambre Y, Flamez D, Drucker DJ, Pipeleers DG & Schuit FC 2001 Glucagon-like peptide 1 receptor signaling influences topography of islet cells in mice. Virchows Archiv 438 382–387.[CrossRef][Web of Science][Medline]
Liu J-L, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U & Liu YL 2004 Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. American Journal of Physiology. Endocrinology and Metabolism 287 E405–E413.
Lu Y, Herrera PL, Guo Y, Sun D, Tang Z, LeRoith D & Liu JL 2004 Pancreatic-specific inactivation of IGF-I gene causes enlarged pancreatic islets and significant resistance to diabetes. Diabetes 53 3131–3141.
Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LIS, Bulman MP, Ayres S, Shepherd M, Clark P, Millward A et al. 1999 Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. Journal of Clinical Investigation 104 R33–R39.[Web of Science][Medline]
Martin J, Hunt SL, Dubus P, Sotillo R, Nehme-Pelluard F, Magnuson MA, Parlow AF, Malumbres M, Ortega S & Barbacid M 2003 Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene 22 5261–5269.[CrossRef][Web of Science][Medline]
Matsuoka TA, Artner I, Henderson E, Means A, Sander M & Stein R 2004 The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. PNAS 101 2930–2933.
Melloul D, Marshak S & Cerasi E 2002 Regulation of pdx-1 gene expression. Diabetes 51 S320–S325.
Montanya E, Nacher V, Biarnes M & Soler J 2000 Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: role of beta-cell hyperplasia and hypertrophy. Diabetes 49 1341–1346.[Abstract]
Naya F, Stellrecht C & Tsai M 1995 Tissue-specific regulation of the insulin gene by a novel basic helix–loop–helix transcription factor. Genes and Development 9 1009–1019.
Naya FJ, Huang H-P, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB & Tsai M-J 1997 Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD-deficient mice. Genes and Development 11 2323–2334.
Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL & Wright CV 1996 PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 122 983–995.[Abstract]
Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A & Accili D 2006 Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. Journal of Clinical Investigation 116 775–782.[CrossRef][Web of Science][Medline]
Olbrot M, Rud J, Moss LG & Sharma A 2002 Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. PNAS 99 6737–6742.
Otani K, Kulkarni RN, Baldwin AC, Krutzfeldt J, Ueki K, Stoffel M, Kahn CR & Polonsky KS 2004 Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. American Journal of Physiology. Endocrinology and Metabolism 286 E41–E49.
Pamir N, Lynn FC, Buchan AMJ, Ehses J, Hinke SA, Pospisilik JA, Miyawaki K, Yamada Y, Seino Y, McIntosh CHS et al. 2003 Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. American Journal of Physiology. Endocrinology and Metabolism 284 E931–E939.
Pei X-H, Bai F, Tsutsui T, Kiyokawa H & Xiong Y 2004 Genetic evidence for functional dependency of p18Ink4c on Cdk4. Molecular and Cellular Biology 24 6653–6664.
Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, Klumperman J, Thorens B & Thomas G 2000 Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 408 994–997.[CrossRef][Medline]
Petrik J, Pell JM, Arany E, McDonald TJ, Dean WL, Reik W & Hill DJ 1999 Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology 140 2353–2363.
Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S & Polonsky K 1998 Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 47 358–364.[Abstract]
Pictet RL, Clark WR, Williams RH & Rutter WJ 1972 An ultrastructural analysis of the developing embryonic pancreas. Developmental Biology 29 436–467.[CrossRef][Web of Science][Medline]
Porter SE, Sorenson RL, Dann P, Garcia-Ocaña A, Stewart AF & Vasavada RC 1998 Progressive pancreatic islet hyperplasia in the islet-targeted, parathyroid hormone-related protein-overexpressing mouse. Endocrinology 139 3743–3751.
Prasadan K, Daume E, Preuett B, Spilde T, Bhatia A, Kobayashi H, Hembree M, Manna P & Gittes GK 2002 Glucagon is required for early insulin-positive differentiation in the developing mouse pancreas. Diabetes 51 3229–3236.
Rajagopal J, Anderson WJ, Kume S, Martinez OI & Melton DA 2003 Insulin staining of ES cell progeny from insulin uptake. Science 299 363.
Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB & Cornelius JG 2000 Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nature Medicine 6 278–282.[CrossRef][Web of Science][Medline]
Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP & Barbacid M 1999 Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nature Genetics 22 44–52.[CrossRef][Web of Science][Medline]
Reusens B & Remacle C 2006 Programming of the endocrine pancreas by the early nutritional environment. International Journal of Biochemistry and Cell Biology 38 913–922.[CrossRef][Web of Science][Medline]
Rhodes CJ 2005 Type 2 diabetes – a matter of beta-cell life and death? Science 307 380–384.
Roccisana J, Reddy V, Vasavada RC, Gonzalez-Pertusa JA, Magnuson MA & Garcia-Ocaña A 2005 Targeted inactivation of hepatocyte growth factor receptor c-met in beta-cells leads to defective insulin secretion and GLUT-2 downregulation without alteration of beta-cell mass. Diabetes 54 2090–2102.
Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A & German M 2000 Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 127 5533–5540.[Abstract]
Scaglia L, Smith F & Bonner-Weir S 1995 Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology 136 5461–5468.[Abstract]
Scaglia L, Cahill CJ, Finegood DT & Bonner-Weir S 1997 Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 138 1736–1741.
Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD & German MS 2000 Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 127 3533–3542.[Abstract]
Smith SB, Gasa R, Watada H, Wang J, Griffen SC & German MS 2003 Neurogenin3 and hepatic nuclear factor 1 cooperate in activating pancreatic expression of Pax4. Journal of Biological Chemistry 278 38254–38259.
Sone H & Kagawa Y 2005 Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 48 58–67.[CrossRef][Web of Science][Medline]
Sosa-Pineda B, Chowdhury K, Torres M, Oliver G & Gruss P 1997 The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 386 399–402.[CrossRef][Medline]
Spooner BS, Walther BT & Rutter WJ 1970 The development of the dorsal and ventral mammalian pancreas in vivo and in vitro. Journal of Cell Biology 47 235–246.
Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J, Magnuson MA & Wu H 2006 Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ-induced diabetes. Molecular and Cellular Biology 26 2772–2781.
Stoffers DA, Ferrer J, Clarke WL & Habener JF 1997a Early-onset type-ll diabetes mellitus (MODY4) linked to IPF1. Nature 17 138–139.
Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL & Habener JF 1997b Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nature Genetics 15 106–110.[CrossRef][Web of Science][Medline]
Sussel L, Kalamaras J, Hartigan-O’Connor DJ, Meneses JJ, Pedersen RA, Rubenstein JL & German MS 1998 Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 125 2213–2221.[Abstract]
Teta M, Long SY, Wartschow LM, Rankin MM & Kushner JA 2005 Very slow turnover of beta-cells in aged adult mice. Diabetes 54 2557–2567.
Tuttle RL, Gill NS, Pugh W, Lee J-P, Koeberlein B, Furth EE, Polonsky KS, Naji A & Birnbaum MJ 2001 Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nature Medicine 7 1133–1137.[CrossRef][Web of Science][Medline]
Uchida T, Nakamura T, Hashimoto N, Matsuda T, Kotani K, Sakaue H, Kido Y, Hayashi Y, Nakayama KI, White MF et al. 2005 Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nature Medicine 11 175–182.[CrossRef][Web of Science][Medline]
Vasavada RC, Garcia-Ocaña A, Zawalich WS, Sorenson RL, Dann P, Syed M, Ogren L, Talamantes F & Stewart AF 2000 Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. Journal of Biological Chemistry 275 15399–15406.
Vasavada RC, Cozar-Castellano I, Sipula D & Stewart AF 2006 Conditional knockout of the retinoblastoma (pRb) gene in beta cells has no effect on beta cell mass or function. Diabetes 55 A364.
Wang Q & Brubaker PL 2002 Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia 45 1263–1273.[CrossRef][Web of Science][Medline]
Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, Schmidt EV & Brand SJ 1993 Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. Journal of Clinical Investigation 92 1349–1356.[Web of Science][Medline]
Williams BO, Remington L, Albert DM, Mukai S, Bronson RT & Jacks T 1994 Cooperative tumorigenic effects of germline mutations in Rb and p53. Nature Genetics 7 480–484.[CrossRef][Web of Science][Medline]
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI et al. 1998 Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391 900–904.[CrossRef][Medline]
Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL & White MF 1999 Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nature Genetics 23 32–40.[Web of Science][Medline]
Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K & Cavener DR 2002 The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Molecular and Cellular Biology 22 3864–3874.
Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K et al. 2005a MafA is a key regulator of glucose-stimulated insulin secretion. Molecular and Cellular Biology 25 4969–4976.
Zhang X, Gaspard JP, Mizukami Y, Li J, Graeme-Cook F & Chung DC 2005b Overexpression of cyclin D1 in pancreatic beta-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes 54 712–719.
Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, Costa RH & Gannon M 2006 The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Molecular Endocrinology 20 1853–1866.
Received in final form 10 November 2006
Accepted 4 December 2006
Made available online as an Accepted Preprint 12 December 2006
This article has been cited by other articles:
 |
R. Granata, M. Volante, F. Settanni, C. Gauna, C. Ghe, M. Annunziata, B. Deidda, I. Gesmundo, T. Abribat, A.-J. van der Lely, et al. Unacylated ghrelin and obestatin increase islet cell mass and prevent diabetes in streptozotocin-treated newborn rats J. Mol. Endocrinol., July 1, 2010; 45(1): 9 - 17. [Abstract] [Full Text] [PDF] |
||||
 |
D. Gupta, M. Peshavaria, N. Monga, T. L. Jetton, and J. L. Leahy Physiologic and Pharmacologic Modulation of Glucose-Dependent Insulinotropic Polypeptide (GIP) Receptor Expression in {beta}-Cells by Peroxisome Proliferator-Activated Receptor (PPAR)-{gamma} Signaling: Possible Mechanism for the GIP Resistance in Type 2 Diabetes Diabetes, June 1, 2010; 59(6): 1445 - 1450. [Abstract] [Full Text] [PDF] |
||||
|
|
J. M. Gregory, J. S. Lilley, A. A. Misfeldt, D. L. Buscariollo, W. E. Russell, and D. J. Moore Incorporating Type 1 Diabetes Prevention Into Clinical Practice Clin. Diabetes, March 1, 2010; 28(2): 61 - 70. [Abstract] [Full Text] [PDF] |
||||
|
|
L. van Burck, A. Blutke, S. Kautz, B. Rathkolb, M. Klaften, S. Wagner, E. Kemter, M. Hrabe de Angelis, E. Wolf, B. Aigner, et al. Phenotypic and pathomorphological characteristics of a novel mutant mouse model for maturity-onset diabetes of the young type 2 (MODY 2) Am J Physiol Endocrinol Metab, March 1, 2010; 298(3): E512 - E523. [Abstract] [Full Text] [PDF] |
||||
|
|
O. Tavana, N. Puebla-Osorio, M. Sang, and C. Zhu Absence of p53-Dependent Apoptosis Combined With Nonhomologous End-Joining Deficiency Leads to a Severe Diabetic Phenotype in Mice Diabetes, January 1, 2010; 59(1): 135 - 142. [Abstract] [Full Text] [PDF] |
||||
|
|
H. S. Kang, Y.-S. Kim, G. ZeRuth, J. Y. Beak, K. Gerrish, G. Kilic, B. Sosa-Pineda, J. Jensen, J. Foley, and A. M. Jetten Transcription Factor Glis3, a Novel Critical Player in the Regulation of Pancreatic {beta}-Cell Development and Insulin Gene Expression Mol. Cell. Biol., December 15, 2009; 29(24): 6366 - 6379. [Abstract] [Full Text] [PDF] |
||||
|
|
L. D. Norquay, K. E. D'Aquino, L. M. Opare-Addo, A. Kuznetsova, M. Haas, J. A. Bluestone, and M. F. White Insulin Receptor Substrate-2 in {beta}-Cells Decreases Diabetes in Nonobese Diabetic Mice Endocrinology, October 1, 2009; 150(10): 4531 - 4540. [Abstract] [Full Text] [PDF] |
||||
|
|
V. Chesnokova, C. Wong, S. Zonis, A. Gruszka, K. Wawrowsky, S.-G. Ren, A. BenShlomo, and R. Yu Diminished Pancreatic {beta}-Cell Mass in Securin-Null Mice Is Caused by {beta}-Cell Apoptosis and Senescence Endocrinology, June 1, 2009; 150(6): 2603 - 2610. [Abstract] [Full Text] [PDF] |
||||
|
|
M. M. Rankin and J. A. Kushner Adaptive {beta}-Cell Proliferation Is Severely Restricted With Advanced Age Diabetes, June 1, 2009; 58(6): 1365 - 1372. [Abstract] [Full Text] [PDF] |
||||
|
|
A. Rafacho, T. M. Cestari, S. R. Taboga, A. C. Boschero, and J. R. Bosqueiro High doses of dexamethasone induce increased {beta}-cell proliferation in pancreatic rat islets Am J Physiol Endocrinol Metab, April 1, 2009; 296(4): E681 - E689. [Abstract] [Full Text] [PDF] |
||||
|
|
L. A. Crawford, M. A. Guney, Y. A. Oh, R. A. DeYoung, D. M. Valenzuela, A. J. Murphy, G. D. Yancopoulos, K. M. Lyons, D. R. Brigstock, A. Economides, et al. Connective Tissue Growth Factor (CTGF) Inactivation Leads to Defects in Islet Cell Lineage Allocation and {beta}-Cell Proliferation during Embryogenesis Mol. Endocrinol., March 1, 2009; 23(3): 324 - 336. [Abstract] [Full Text] [PDF] |
||||
|
|
C. Broca, J. Quoyer, S. Costes, N. Linck, A. Varrault, P.-M. Deffayet, J. Bockaert, S. Dalle, and G. Bertrand {beta}-Arrestin 1 Is Required for PAC1 Receptor-mediated Potentiation of Long-lasting ERK1/2 Activation by Glucose in Pancreatic {beta}-Cells J. Biol. Chem., February 13, 2009; 284(7): 4332 - 4342. [Abstract] [Full Text] [PDF] |
||||
|
|
A. Ackermann Misfeldt, R. H. Costa, and M. Gannon {beta}-Cell Proliferation, but Not Neogenesis, Following 60% Partial Pancreatectomy Is Impaired in the Absence of FoxM1 Diabetes, November 1, 2008; 57(11): 3069 - 3077. [Abstract] [Full Text] [PDF] |
||||
|
|
J. Clements, K. Hens, C. Francis, A. Schellens, and P. Callaerts Conserved role for the Drosophila Pax6 homolog Eyeless in differentiation and function of insulin-producing neurons PNAS, October 21, 2008; 105(42): 16183 - 16188. [Abstract] [Full Text] [PDF] |
||||
|
|
M. V. Chakravarthy, Y. Zhu, M. B. Wice, T. Coleman, K. L. Pappan, C. A. Marshall, M. L. McDaniel, and C. F. Semenkovich Decreased Fetal Size Is Associated With {beta}-Cell Hyperfunction in Early Life and Failure With Age Diabetes, October 1, 2008; 57(10): 2698 - 2707. [Abstract] [Full Text] [PDF] |
||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |