| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
|
|
||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
REVIEW |
1 Section of General Pathology, Department of Biomedical Sciences
2 Section of Endocrinology, Department of Internal and Specialistic Medicine, Garibaldi-Nesima Hospital, University of Catania, 95122 Catania, Italy
(Correspondence should be addressed to R Vigneri; Email: vigneri{at}unict.it)
| Abstract |
|---|
| Introduction |
|---|
The association between cancer and diabetes has been investigated extensively and most, but not all studies, found that DM is associated with an increased risk of several types of cancer. Most published data, however, requires reinterpretation because DM is not a single disease, but rather a group of metabolic disorders characterized by hyperglycemia. Within this general context, each type of diabetes has additional metabolic and hormonal abnormalities that differently affect diabetic patients. It is therefore inappropriate to consider diabetic patients as a homogeneous cohort. In addition, a series of potential confounders directly related to the disease (obesity, quality of metabolic control, drugs employed for treatment, diet, etc.) and present in diabetic patients may influence the association between diabetes and cancer.
In the present review, we will discuss the available evidence concerning the association between diabetes and cancer, the different aspects of diabetes which may influence this association, and the possible mechanisms involved.
| Cancer risk is increased in diabetic patients |
|---|
|
The incidence of liver and pancreatic cancer is increased in diabetes
Several meta-analyses indicate that the strongest association between DM and increased cancer risk is with pancreatic and liver cancer (Table 1), i.e. two key organs involved in the metabolic derangements typical of diabetes.
Because of the portal circulation, liver cells are exposed to higher insulin concentrations than other tissues, a condition that is exacerbated in insulin-resistant hyperinsulinemic type 2 diabetic individuals, but that is not present in insulin-deficient type 1 diabetic patients treated with exogenous insulin (see Fig. 1). It is unlikely, therefore, that insulin's mitogenic action is specifically involved in the higher incidence of liver cancer in diabetic patients since healthy liver cells are physiologically exposed to higher insulin concentrations than other tissues. Moreover, in diabetic patients injected with exogenous insulin, the liver is exposed to the same insulin levels as the other organs.
|
In conclusion, increased liver cancer incidence in diabetes is well documented and, although the exact mechanisms underlying this association are sill unclear, liver inflammation, hepatocyte damage, and repair are likely to be involved in the higher frequency of HCC among diabetic patients.
Most earlier studies investigating the association between diabetes and pancreatic cancer are probably misleading because they do not distinguish between pre-existing diabetes (a condition possibly favoring exocrine pancreatic cancer) and new-onset diabetes (a possible sign of pancreatic functional damage due to a still undiagnosed pancreatic cancer; Noy & Bilezikian 1994). The latter situation is so frequent that hyperglycemia and diabetes, when appearing after the age of 45-50 years, in a lean subject with no family history for diabetes, is considered sufficient to pose an indication for pancreatic cancer screening (Noy & Bilezikian 1994, Chari et al. 2008, Pannala et al. 2009). Similarly, elderly subjects with new-onset diabetes have a 3-year risk of pancreatic cancer nearly eight times higher than a nondiabetic person of similar age and sex (Chari et al. 2005). Laboratory and clinical evidence suggest that diabetes caused by pancreatic cancer is due to cytokines produced by the tumor (Basso et al. 2002) rather than secondary to endocrine pancreatic tissue invasion and damage (Pannala et al. 2009). This conclusion is also supported by the observation that hyperglycemia occurs at an early stage of pancreatic cancer and is independent of tumor size and stage (Chari et al. 2008, Pannala et al. 2008). Epidemiological studies in subjects affected by DM at least 1 year prior to diagnosis or death from pancreatic cancer indicated a relative risk (RR) of 2.1 (95% confidence interval (CI)=1.6-2.8). When the same analysis was carried out including only patients with 5 years of pre-diagnosed diabetes, their RR for pancreatic cancer was similar (RR=2.0; Everhart & Wright 1995). Since all of these data exclude diabetes induced by pancreatic tumors, the reported findings support the possibility that diabetes is indeed a risk factor for pancreatic cancer.
The ‘pre-diabetes’ state should also be considered a risk factor for pancreatic cancer. Studies that evaluated the association between post-load glucose levels and pancreatic tumors in 35 658 individuals reported a higher RR with increasing glucose tolerance impairment. After adjusting for age, race, cigarette smoking, and body mass index (BMI), the risk progressively increased from normal subjects to subjects with slightly altered glycemia (RR=1.65) and then to diabetes (RR=2.15; Gapstur et al. 2000). These results did not change when patients who died of pancreatic cancer during the first 5 years after the assessment of post-load glucose levels were excluded, further suggesting that hyperglycemia and diabetes per se are predisposing factors for pancreatic cancer.
The biological mechanisms underlying the association between diabetes and pancreatic cancer are unclear. Hyperinsulinemia has been indicated as a possible factor because exocrine pancreatic cells, which give rise to most pancreatic cancers, are exposed to very high insulin concentrations because of the common blood supply with the adjacent insulin-secreting islets (Williams & Goldfine 1985). Elevated insulin could act as a tumor growth-promoting factor in many different ways (covered later). This mechanism, however, does not justify the excess of pancreatic cancer in insulin-treated diabetic patients (Green & Jensen 1985) or in type 1 diabetes (Stevens et al. 2007) where pancreatic cells are not exposed to insulin levels higher than those of other tissues. In these studies, however, the analysis is hampered by the insufficient number of cases accrued, due to both the type of diabetes (type 1 diabetes accounts for <10% of all DM patients) and patient age (pancreatic cancer is rare before age 40).
Increased incidence of other cancers in diabetes
An increased frequency of malignancies of other organs has been reported in diabetic patients and has been ascribed to a variety of general and local mechanisms. In these cases, studies are not as numerous as for liver and pancreatic tumors, and the increases in RR are not as statistically significant. However, in many instances, the increased risk is clinically relevant, especially considering the prevalence of the two diseases in the general population.
In diabetic patients, the increased incidence and increased mortality for kidney cancer have been attributed to both general mechanisms (hyperinsulinemia and obesity) and specific factors, mainly hypertension (Yuan et al. 1998, Chow et al. 2000, Zucchetto et al. 2007) and the frequent kidney diseases occurring in diabetic patients (Lindblad & Adami 2002).
Individuals with DM also display a modest increase in the risk of bladder cancer. In this case, in addition to general factors like hyperinsulinemia, the increased frequency of urinary tract infections is also likely to be involved.
The risk of cancers of the female reproductive organs is also increased in DM. Both breast and endometrial cancer risks are increased in diabetic women, and this risk is independent from obesity (a well-established factor promoting breast cancer) as it persists even after correcting epidemiological data for this disease.
Several biological mechanisms may be involved, mostly regarding sex hormone abnormalities. Hyperinsulinemia may increase the levels of bioactive estrogens by decreasing the concentration of circulating sex hormone-binding globulin (Kaaks 1996) and might also stimulate androgen synthesis in the ovarian stroma (Kaaks 1996). Other possible mechanisms include delayed menarche, especially in type 1 diabetic women, who also have a higher incidence of nulliparity, irregular menses, and fertility disorders.
Type 2 diabetes has been associated with an increased risk of colorectal adenomas and carcinomas in most, but not all, studies (Elwing et al. 2006, Limburg et al. 2006). The risk is increased in both women and men for both colon and rectal cancer (Larsson et al. 2005). In addition to hyperinsulinemia, hypothesized mechanisms include slower bowel transit time and the elevated fecal bile acid concentrations often observed in DM (Stadler et al. 1988, Will et al. 1998).
Large prospective cohort studies and case-control studies have shown a moderate increase of non-Hodgkin's lymphoma in diabetic patients, a possible consequence of the immune dysfunction related to impaired neutrophil activity and abnormalities in cellular and humoral immunity in diabetes (Mitri et al. 2008).
Decreased incidence of prostate cancer in diabetes
In contrast to the increased risk of numerous forms of neoplasia, most studies report a reduced risk of prostate cancer in men with diabetes. A recent meta-analysis (Kasper & Giovannucci 2006) including 14 studies carried out in the pre-PSA era (i.e. before the generalised use of prostate specific antigen screening for prostate cancer; Bonovas et al. 2004) and 5 additional studies carried out in the PSA era (and therefore, concerning cancers diagnosed earlier and smaller cancers) has found a significantly reduced risk in diabetic patients (Table 1). The 16% average decreased risk of developing prostate cancer must most likely be attributed to the decreased testosterone levels in diabetic patients (Barrett-Connor 1992, Betancourt-Albrecht & Cunningham 2003). However, other metabolic and hormonal factors, including altered insulin and leptin concentrations, the diffuse use of medications such as statins and metformin, and changes in diet and lifestyle in order to control diabetes, have also been hypothesized as elements potentially contributing to the inverse association between diabetes and prostate cancer (Kasper & Giovannucci 2006).
In conclusion, the epidemiological studies cited above may be partially biased by relevant heterogeneity due to different study design (inclusion criteria), incomplete characterization of DM, failure to consider potential confounders (obesity, diabetes duration, and treatment), and also variably defined control population. However, the overall increased risk for the development of several types of cancer in diabetic patients must be considered well documented. In diabetes, there is a mild to moderate increase in the incidence of pancreas, liver, breast, colorectal, urinary tract, and female reproductive organ cancer and a mild reduction in prostate cancer risk.
| Cancer mortality is increased in diabetic patients |
|---|
A positive association between breast cancer mortality and diabetes was found in three out of five studies, with a RR from the pooled data of the five studies of 1.24 (95% CI=0.95-1.62; Larsson et al. 2007). In the largest study (cohort size 588 321 with 4346 deaths from breast cancer), after adjusting for age, race, BMI, physical activity, smoking, and alcohol, RR in diabetic women was 1.27 (1.11-1.45) when compared with the nondiabetic female population. In this cohort, as in most others, no stratification was performed for type of diabetes and different treatments. In addition, the menopausal status was not recorded (Coughlin et al. 2004). In a recent study aimed at evaluating whether diabetes could affect breast cancer prognosis, after a 5-year mean follow-up, mortality for breast cancer was significantly higher in women with diabetes (hazard ratio 1.39; 95% CI=1.22-1.59, P<0.0001) suggesting that early survival following breast cancer was reduced in women with diabetes (Lipscombe et al. 2008). This reduced survival might be a consequence of more aggressive breast cancer but also of diabetes-related comorbidities. In fact, in that study, the cause of death was not recorded and diabetic women without breast cancer had an increase in mortality similar to that of diabetic women with breast cancer, suggesting that diabetes, rather than breast cancer, was the major factor contributing to the raised mortality.
Diabetes was also positively associated with colorectal cancer mortality. A statistically significant association was found in three out of six studies (Larsson et al. 2005), and a nonsignificant positive association was reported in a fourth one. Pooled data from the six studies indicated a positive association between diabetes and colorectal cancer mortality (RR=1.26; 95% CI=1.05-1.50), but heterogeneity issues partially compromise the significance of the results. Within these six articles, the two cohort studies that evaluated standardized mortality ratio both indicated a positive association between DM and colorectal cancer death. However, only one study reported a statistically significant increased mortality from colorectal cancer in diabetic patients. A study aimed at evaluating the influence of diabetes on long-term outcome of patients resected for colon cancer (3759 patients, 287 with DM) found that diabetes negatively affected survival in colon cancer patients (Meyerhardt et al. 2003). Data were adjusted for predictors of colon cancer outcome (age, gender, race, clinical status, TNM (tumor, node, metastasis classification) category, Dukes stage, location of primary tumor, and grade of differentiation), and indicated that both disease-free survival (DFS) and overall survival (OS) at 5 years were significantly reduced in diabetic patients (DFS=48 vs 59% in nondiabetics, P<0.0001; OS=57 vs 66% in nondiabetics, P<0.0001). Median survival in diabetic patients was 6.0 years vs 11.3 in nondiabetic subjects. In this study, the role of DM comorbidities (that may negatively affect overall mortality among cancer patients because of adverse health conditions) was probably minor since cancer recurrence was also higher in diabetic patients (recurrence-free survival 56 vs 64% in nondiabetics, P<0.012).
A positive association was also found between diabetes and endometrial cancer mortality in two studies, but it was significant only in one of them (RR=2.38; 95% CI=1.05-5.37; Coughlin et al. 2004, Folsom et al. 2004).
It is interesting to note that, although diabetic patients have a reduced risk of prostate cancer, once an insulin-resistant, overweight man has been diagnosed with prostate cancer, his likelihood of dying from the disease is increased (Ma et al. 2008).
A recent study on the systematic assessment of long-term, all-cause mortality in cancer patients with or without diabetes has evaluated, at 1.41 (95% CI=1.28-1.55), the hazard ratio for death in cancer patients with diabetes compared to cancer patients without diabetes (Barone et al. 2008). Mortality was significantly increased for cancers of the breast, endometrium, colon, and rectum. In this study, the increase in mortality risk was not significantly increased for lung, gastric, liver, pancreatic or prostate cancers. Overall, however, the heterogeneity of the studies analyzed and the length of the observation period (1969-2008, during which treatment for both cancer and diabetes changed markedly) hamper, at least in part, the significance of the data.
Several possible explanations can be put forth to explain the increased risk of cancer death in DM. For instance, it is still unclear whether diabetes, through a number of mechanisms, makes the cancer more aggressive or whether the host organism is less resistant to cancer progression. It is also possible that diabetic patients receive different cancer treatment (i.e. oncologists may employ lower chemotherapy doses in diabetic patients, concerned about their general health and their heart, liver, and kidney function). Of course, it is also possible that diabetic patients may have a worse response to chemotherapy compared with nondiabetic individuals.
In conclusion, epidemiologic studies provide evidence that cancer mortality is moderately increased in diabetic patients. Whether this is a consequence of hyperglycemia and hyperinsulinemia (growth-promoting effect on cancer cells), the impaired health conditions due to diabetes' comorbidities or a combination of the two is still unclear.
| Type 1 and type 2 diabetes and cancer risk |
|---|
In type 2 diabetes, hyperglycemia and hyperinsulinemia coexist for a long time because of insulin resistance in peripheral tissues. Only when β-cell function fails completely will the patient require insulin treatment because of endogenous insulin deficiency.
In spite of these considerable pathogenetic and clinical differences, many studies on the association between diabetes and cancer were carried out without an appropriate distinction between the two forms of diabetes.
For obvious epidemiological reasons, most studies on the association between cancer and diabetes have been carried out in patients with type 2 diabetes (90% of all diabetic patients). As these patients, unlike those with type 1 diabetes, have endogenous hyperinsulinemia and insulin resistance, it is questionable whether these data can be automatically extended to type 1 diabetic patients. This concern is particularly relevant for the older reports in which diabetes assessment was based on self-reported hyperglycemia, with no criteria aimed at distinguishing type 1 from type 2 diabetes. Although more recent studies have been based on medical records, the distinction between type 1 and type 2 diabetes was mostly based on surrogate indicators of diabetes type, like young patient age or insulin treatment (assumed as type 1) versus insulin-independent diabetes (assumed as type 2). This distinction does not take into account many specific conditions, including type 2 diabetic patients that are treated with insulin because oral hypoglycemic agents (OHA) are no longer effective (secondary failure to OHA), type 1 diabetes of the adult (5% of adult subjects previously classified as type 2 diabetes; Buzzetti et al. 2007), and other less frequent conditions.
Because of the 10:1 ratio between type 2 and type 1 diabetes, and considering that cancer is mainly a disease of the older population (where type 1 diabetes is less frequent), it is reasonable to assume that the large majority of tumors observed in diabetic patients occurred in type 2 diabetics.
Thus, if cancer association with type 1 diabetes has specific characteristics, these have likely been obscured by the large majority of cancers diagnosed in type 2 diabetic patients.
Even the few studies specifically addressing cancer incidence in type 1 diabetic patients suffer from poor diabetes type assessment. For example, a recent cohort study evaluating cancer incidence in nearly 30 000 Swedish type 1 diabetic patients diagnosed in the period 1965-1999 has identified 355 cases of cancer (standardized incidence ratio (SIR)=1.2; 95% CI=1.0-1.3, compared with the general Sweden population; Zendehdel et al. 2003). In contrast to type 2 diabetic patients, no increased risk of breast, pancreatic, colorectal, or kidney cancer was found in this cohort. However, type 1 diabetic patients had an increased RR for stomach (SIR=2.3; 95% CI=1.1-4.1), endometrial (SIR=2.7; 95% CI=1.4-4.7), and cervical cancer (1.6; 1.1-2.2). These positive associations have been attributed to the high prevalence of Helicobacter pylori infection or of pernicious anemia (for gastric carcinomas; Oldenburg et al. 1996, De Block et al. 1999) and to the higher incidence of nulliparity, irregular menses, and fertility disorders in type 1 diabetic women (for uterine malignancies). In contrast with this report, a recent meta-analysis including three cohort studies and six case-control studies has found that the RR for pancreatic cancer was doubled in type 1 diabetic patients and young-onset diabetics in comparison with nondiabetics (Stevens et al. 2007).
In conclusion, the large majority of the epidemiological data on cancer incidence and mortality has been obtained in type 2 diabetic patients. Because of the different biology between the two subtypes of diabetes, these findings cannot be acritically extended to type 1 diabetic subjects.
| The role of hyperinsulinemia in favoring cancer incidence and progression in diabetic patients |
|---|
Most type 1 and type 2 diabetic patients are exposed for decades to increased insulin concentrations, although the physiologic and therapeutic conditions are very different in each individual with diabetes.
Type 1 diabetic patients have an absolute requirement for exogenous insulin because of autoimmune destruction of their pancreatic β-cells, which are therefore unable to produce endogenous insulin. In these patients, insulin administration cannot mimic the physiologic insulin secretion, not only in terms of temporal pattern and hormone serum levels but also in terms of compartment distribution. Indeed, pancreas-secreted insulin is first distributed to the liver (first passage insulin) where a relevant aliquot (up to 80%; Ferrannini & Cobelli 1987) is retained and degraded. The remaining hormone reaches the peripheral tissues through the systemic circulation. The liver/peripheral tissue insulin concentration ratio, therefore, ranges from 3:1 up to 9:1 during insulin secretion bursts. Exogenously administered insulin, in contrast, will arrive to peripheral tissues and to the liver at the same time and at a similar concentration. Peripheral tissue hyperinsulinemia due to exogenous insulin (circulating levels may peak two- to fivefold higher than normal endogenous levels, depending on the dose injected and the type of insulin or analog used) and the ensuing relative liver hypoinsulinemia, therefore, are a common condition in type 1 diabetic patients (Fig. 2).
|
As DM persists for many years, this scenario is often subject to changes, with most type 2 diabetic patients progressively presenting decreased insulin secretion following the failure of β-cells, due to increased apoptosis rates that are not balanced by neogenesis. At this stage, patients with type 2 diabetes may become similar to type 1 diabetic individuals, with endogenous hypoinsulinemia and exogenous insulin requirement.
When studying type 2 diabetic patients, therefore, diabetes duration and insulin requirement may affect tissue exposure to insulin in different ways. If hyperinsulinemia has a role in promoting cancer initiation and/or progression, these aspects should be considered when determining the individual risk of a diabetic patient to develop cancer. Most studies on the diabetes-cancer association overlooked these different biological conditions.
In conclusion, diabetes is generally characterized by hyperglycemia and hyperinsulinemia, often coupled with a reduced metabolic effect of insulin (insulin resistance) in peripheral tissues. Chronic hyperinsulinemia, however, is a possible factor favoring cancer initiation and/or progression in diabetic patients due to the mitogenic effect of insulin. The heterogeneity and complexity of different tissue exposure to hyperinsulinemia in diabetic individuals does not allow the quantification of the role of insulin in promoting cancer risk in the different organs of different diabetic patients.
One example is the potentially increased risk of lung cancer in diabetic patients using the recently introduced inhaled insulin (von Kriegstein & von Kriegstein 2007). The long-term effects of this form of therapy are unknown. Although short-term studies in animals have shown no substantial effect on cell proliferation indices, the high insulin concentration at alveolar and bronchiolar epithelia (due to the fact that only 10-25% of inhaled insulin is absorbed) has raised safety concerns about the possibility that it may promote lung cancer. These concerns have been recently reinforced by the long-time surveillance analysis, indicating that 6 out of 4740 (0.13%) diabetic patients treated with inhaled insulin but only 1 out of 4292 comparator-treated patients (0.02%) developed lung cancer (Mitri & Pittas 2009).
There are multiple and complex mechanisms potentially responsible for the mitogenic effects of insulin.
First, when insulin levels increase (as in the post-prandial surge in insulin-resistant subjects or after insulin injection), insulin may bind and activate the related insulin-like growth factor-I (IGF-I) receptor, which shares 80% homology with the insulin receptor (IR), but has a more potent mitogenic and transforming activity. Moreover, insulin decreases IGF-I-binding proteins (IGF-BP1 and, perhaps, IGF-BP2; Kaaks & Lukanova 2001): this will result in increased free IGF-I, the biologically active form of the growth factor.
Secondly, many cancer cells have an increased IR content (Papa et al. 1990; Fig. 3A). The IR may be expressed in two different isoforms, A and B, produced by an alternative splicing of the IR gene transcript (Moller et al. 1989). In malignant cells, the A isoform (IR-A) expression is predominant (Frasca et al. 1999, Sciacca et al. 1999, Kalli et al. 2002; Fig. 3B), and its activation, at variance with the IR-B isoform, elicits more mitogenic than metabolic effects (Frasca et al. 1999). By binding to the overexpressed IR-A, insulin may favor cancer progression and facilitate the growth of tumors that would otherwise have likely remained clinically irrelevant for an undetermined length of time.
|
|
| Anti-diabetic drugs that may influence cancer risk in diabetic patients |
|---|
|
Recent studies in MCF-7, BT-474, and SKBR-3 human breast cancer cells showed that in vitro metformin inhibited cell proliferation, reduced colony formation, and caused partial cell cycle arrest (Alimova et al. 2009). These effects mainly occurred via MAPK, AKT, and mTOR inhibition and were replicated also in erbB2-overexpressing cells. On the basis of both epidemiological data and in vitro studies, a clinical trial for evaluating metformin activity on breast cancer cell proliferation (Ki67 index) is currently undergoing in 100 breast cancer patients (Cazzaniga et al. 2009).
Data on the other insulin-sensitizing drug (thiazolidinediones) are more controversial. A beneficial (Govindarajan et al. 2007), neutral (Koro et al. 2007), or even deleterious (Ramos-Nino et al. 2007) effect has been reported for different types of cancer. The biological mechanism of these compounds is to activate PPAR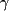 receptors, which, in several in vitro experimental models, has shown a potential anti-cancer effect (Aiello et al. 2006). In addition to lowering hyperinsulinemia, this effect can explain the described anti-cancer effect of glitazones. In any case, the use of these compounds is too recent and too limited to consider the present meager epidemiologic observations reliable.
receptors, which, in several in vitro experimental models, has shown a potential anti-cancer effect (Aiello et al. 2006). In addition to lowering hyperinsulinemia, this effect can explain the described anti-cancer effect of glitazones. In any case, the use of these compounds is too recent and too limited to consider the present meager epidemiologic observations reliable.
The third group of drugs (sulphonylureas) are secretagogues, i.e. increase insulin secretion and cause hyperinsulinemia. As expected, therefore, they have been associated with an increased risk of cancer (Bowker et al. 2006). Different sulphonylureas may have different effects, with glyburide being more deleterious than gliclazide (Monami et al. 2007). Although their effect on cancer risk is attributed to the prolonged hyperinsulinemia that they induce in patients, a direct effect on cancer (either positive or negative) cannot be excluded.
In conclusion, some evidence suggests that the biguanide metformin may reduce cancer risk in diabetic patients but, in general, the influence of anti-diabetic drugs on the risk of cancer is not well studied and evidence is weak, indirect, and controversial.
| Other factors that may influence the risk of cancer in diabetes |
|---|
Over 80% of type 2 diabetic patients are obese. Obesity is associated with a higher incidence and a higher mortality in cancer (Adami & Trichopoulos 2003, Vigneri et al. 2006). Moreover, cancer mortality significantly increases with increasing patient BMI (Calle et al. 2003). Fat distribution in the body is also important: central (upper body or android) obesity is more harmful than gynoid obesity in terms of increased risk and worst cancer outcome. Given these observations, it is evident that studies on the diabetes-cancer association are influenced by the high prevalence of obesity in DM patients. Since both DM and obesity are characterized by hyperinsulinemia and higher cancer incidence, it is difficult to identify the contribution of each of the two conditions.
Among the many possible mechanisms involved, hyperinsulinemia (which is typical of central obesity), diet and nutritional factors causing a positive energy balance, and other hormone abnormalities have been indicated as causal factors.
A tight correlation has been observed between obesity, circulating estrogen levels, and increased breast cancer risk (Key et al. 2003, Cleary & Grossmann 2009) especially in post-menopausal women. Obese post-menopausal women usually present an increase in both estrone and estradiol, a likely consequence of the increased aromatase activity of the adipose tissue (Reed & Purohit 2001). Considering the growing prevalence of obesity and diabetes in both developed and developing countries, these data might explain the reported rise in estrogen receptor-positive breast cancers (Glass et al. 2007). Several other molecular alterations associated with obesity might also be responsible for the higher incidence of breast cancer found in obese (and obese-diabetic) pre- and post-menopausal women. Preclinical evidence has suggested that leptin, an adipocyte-derived cytokine, highly expressed in obese subjects, promotes breast cancer cell proliferation (Hu et al. 2002), an observation that has not yet been confirmed in the clinical setting since an association between leptin levels and breast cancer outcome has not been demonstrated (Goodwin et al. 2005). Another adipokine produced by the adipose tissue, adiponectin, which is inversely correlated with body fat, might exert a protective effect on breast epithelial cells since its addition to different breast cancer cell lines inhibited proliferation and enhanced apoptosis (Cleary et al. 2009).
Hyperglycemia
Most diabetic patients present both hyperglycemia and hyperinsulinemia. Thus, it is difficult to distinguish the specific role of each abnormality in increasing cancer risk.
It is known that a high intake of sugar and refined carbohydrates and elevated blood glucose levels are strongly associated with the risk of cancer (Krone & Ely 2005). It is also known that impaired glucose tolerance without diabetes is associated with increased cancer risk (Dankner et al. 2007). Both these conditions, however, are also characterized by hyperinsulinemia. Although much convincing evidence demonstrates an association between hyperglycemia and cancer, it has yet to be demonstrated that hyperglycemia per se is an independent risk factor.
Possible mechanisms implicated include the role of an abnormal energy balance and the effect of hyperglycemia in impairing the effect of ascorbic acid on the intracellular metabolism and reducing the effectiveness of the immune system. Further evidence suggests a role for the oxidative stress-responsive genes (like thioreodoxin-interacting protein) that are sensitive to hyperglycemia and regulate the level of reactive oxygen species (ROS; Turturro et al. 2007).
Free fatty acids
Deregulation of fatty acid synthase (FASN) activity, which catalyzes de novo fatty acids biogenesis (Hillgartner et al. 1995, Semenkovich et al. 1995), could also play a role in the pathogenesis of insulin resistance, diabetes, and cancer. FASN activity is important for de novo fatty acid synthesis in the liver and is stimulated by a low-fat/high-carbohydrate diet (Hudgins 2000, Hudgins et al. 2000). Interestingly, FASN expression is increased in insulin-resistant/hyperinsulinemic patients (Moustaid et al. 1996, Claycombe et al. 1998), and its increased activity further worsens insulin resistance and may result in NAFLD (Postic & Girard 2008), which is associated with an increased risk of hepatocarcinoma (Caldwell & Lazo 2009). FASN activity is also increased in cancer cells, where de novo fatty acid synthesis is crucial for membrane remodeling during cell migration and proliferation, as well as for lipid-based post-translational modifications of intracellular proteins in highly proliferating cell populations (i.e. myristylation of RAS). The concept that FASN is directly involved in affecting tumor progression derives also from studies with the FASN blocker cerulenin (Lupu & Menendez 2006a,b). Indeed, cell exposure to this inhibitor results in cytostatic, cytotoxic, and apoptotic effects in vitro and retards the growth of tumor in xenograft models (Menendez et al. 2009).
Therefore, FASN activity and fatty acid production are another possible link between diabetes and cancer as indicated by the hypothesis that insulin-resistant conditions such as obesity, type 2 diabetes, and cancer are favored by common FASN-driven ‘lipogenic state’ (Menendez et al. 2009).
Chronic inflammation and oxidative stress
The metabolic abnormalities that characterize diabetes, especially under conditions of poor metabolic control, increase oxidative stress and cause a permanent pro-inflammatory condition. This chronic pro-inflammatory state (which persists for years or decades) reduces intracellular anti-oxidant capacity, predisposing susceptible cells to malignant transformation. In fact, high concentrations of diverse free radicals and oxidants generate a potent ROS that can damage cell DNA by direct oxidation or by interfering with the mechanisms of DNA repair (Federico et al. 2007). ROS may also react with proteins and lipids, forming derivative products that may alter intracellular homeostasis favoring the accumulation of mutations that, in turn, contribute to the multistage carcinogenesis process (Ohshima et al. 2003).
A possible additional mechanism is related to mitochondrial dysfunction, a well-recognized abnormality in diabetes. DNA repair is a high energy consuming process that requires increased mitochondrial activity: stimulating malfunctioning mitochondria will not only provide low, insufficient energy supply, but also increase ROS production (Cebioglu et al. 2008).
Moreover, an additional factor correlated with insulin resistance is the pro-inflammatory cytokine tumor necrosis factor (TNF) produced by the adipose tissue (Kern et al. 2001). TNF induces development and progression of many tumors (Szlosarek et al. 2006) by strongly activating nuclear factor-kappa B (NF-B), which mediates many of the pro-tumoral effects of TNF.
In conclusion, DM, by mechanisms both specific to diabetes and common with other chronic degenerative diseases, might accelerate the aging biological processes that favor cancerogenesis.
| Drugs used to treat cancer may favor diabetes |
|---|
Apart from corticosteroids, anti-androgens may also adversely affect glucose metabolism. Androgen deprivation therapy is the fundamental treatment of prostate cancer. This therapy causes a variety of metabolic abnormalities that include decreased insulin sensitivity and altered lipid profile. It therefore, increases risk of diabetes and cardiovascular disease (Saylor & Smith 2009).
Androgens are important determinants of body composition: their inhibition increases fat mass and decreases lean body mass. In patients treated with GnRH agonists and/or nonsteroidal anti-androgens, such as flutamide, bicalutamide, and nilutamide, or with the steroidal anti-androgen cyproterone acetate, ‘sarcopenic obesity’ is favored, a combination of excess body weight and reduced muscle mass. Fat accumulation is primarily subcutaneous and is often associated with increased total cholesterol, triglycerides, and high-density lipoprotein (HDL). These changes result in insulin resistance and, sometimes, diabetes. In a recent study in over 70 000 subjects with locoregional prostate cancer, those who were treated with GnRH had a 44% increased risk of developing diabetes (Keating et al. 2006). Diet and lifestyle interventions with a 5-10% weight loss and statin drugs are the main strategies for preventing or treating the metabolic complications of androgen deprivation therapy in prostate cancer patients.
The other most currently employed targeted anti-cancer molecules do not significantly affect glucose homeostasis. However, an increasing number of compounds are being tested for therapeutic use which alter the IGF-I system and its intracellular pathways. The increasing use of these compounds may amplify the frequency of anti-cancer drug-related diabetes. Since IGF-I signaling plays a key role in both tumor progression and glucose homeostasis, therapies targeting the IGF system for its pro-cancer effect may at the same time cause hyperglycemia. In this paragraph, we will examine drugs and mechanisms responsible for hyperglycemia induced by novel anti-cancer therapies that may alter the insulin-glucose balance.
IGF-I system targeting anti-cancer treatments
IGF-I and insulin, their receptors and their intracellular signaling pathways share large similarities. Likewise, the biological (metabolic and mitogenic) effects of the two hormones partially overlap. Because of the well-known role of IGF-I as a cancer-promoting factor, many efforts have been made to block its function in cancer patients. However, these efforts may have a detrimental effect on glucose metabolism through three different mechanisms: i) the inhibition of the IGF-I insulin-mimetic effect (Kuzuya et al. 1993, Fernandez et al. 2001, Pennisi et al. 2006); ii) the increase in circulating GH levels due to the lack of IGF-I feedback (GH is a potent diabetogenic hormone; Yakar et al. 2004); and iii) the possibility that agents that block IGF-I signaling might also cross inhibit the insulin-signaling pathway.
Currently, anti-cancer strategies inhibiting the IGF system include both direct targeting of the IGF-I receptor (IGF-IR) with both monoclonal antibodies and suppression of the IGF-IR-signaling pathway by protein kinase inhibitors (Fig. 5).
|
Hyperglycemia has been observed in a few patients enrolled in studies with the anti-IGF-IR antibody (Haluska et al. 2007, Lacy et al. 2008). This is likely to be a consequence of a compensatory increase in the circulating concentration of GH after IGF-I blockade, with the consequent insulin resistance (del Rincon et al. 2007) that may cause or worsen diabetes.
A second approach to IGF-I inhibition is to block IGF-IR signaling at the enzymatic level. Since IGF-IR is a transmembrane tyrosine kinase (TK) receptor, several TK inhibitors targeting IGF-IR have been developed and found to be active in preclinical models and in phase I clinical trials (Hofmann & Garcia-Echeverria 2005, Gable et al. 2006, Haluska et al. 2006, Ji et al. 2007, Mulvihill et al. 2008, Vasilcanu et al. 2008, Zimmermann et al. 2008). These small molecules may cause more serious toxicity than that observed with the IGF-IR-specific antibodies, as they cross the blood-brain barrier with the possibility of neurotoxicity for the inhibition of the neuroprotective effect of IGF-I. Unexpectedly, these TK inhibitors are associated with less hyperglycemia than IGF-IR-blocking antibodies. One possible explanation for this difference is that the TK inhibitors do not accumulate in muscle, leaving unaffected IR function on the metabolic process of this tissue (Pollak 2008). More research is needed to clarify this point.
Downstream of the receptor, IGF-I signaling occurs via the activation of enzymes and substrates like phosphatidylinositol 3-kinase (PI3K), AKT, and mTOR. When activated via IGF-IR, these substrates play a role in tumor cell proliferation and survival, but they are also activated via the IR and heavily contribute to glucose homeostasis. Several compounds targeting different signaling molecules downstream of the IGF-IR have been tested, as anti-cancer therapies are able to inhibit the mitogenic and anti-apoptotic effects of IGF-I in cancer cells (Fig. 5).
Targeting PI3K, the most proximal pathway component, has the advantage of providing a broader inhibition of downstream signaling compared with distal component inhibition (such as AKT and mTOR). Inhibitors like LY294002 and wortmannin effectively inhibit PI3K, but poor solubility and high toxicity have prevented their clinical application. New compounds (like PX-866) are now being tested in xenograft models and in phase I clinical trials (Ihle et al. 2004, 2009a,b, LoPiccolo et al. 2008). In xenograft models, PX-866 increases glucose and insulin levels as well as glucose intolerance. While metformin is not effective in counteracting this effect and lowering glucose levels, glitazones (e.g. pioglitazone) ameliorate glucose balance in these patients, without affecting the anti-tumor activity of the compound (Ihle et al. 2004, 2009a,b, LoPiccolo et al. 2008).
A variety of AKT inhibitors have been developed (including perifosine, phosphatidylinositol ether lipid analogs PIAs, and triciribine phosphate; Ihle et al. 2004, 2009a,b, LoPiccolo et al. 2008; Fig. 5). Clinical data concerning the anti-tumor activity of AKT inhibitors as well as their effect on glucose homeostasis are insufficient. Recent preliminary data obtained in a xenograft model with GSK690693, a novel ATP-competitive/pan-AKT kinase inhibitor, indicate that abrogating AKT activity results in increased glucose and insulin levels. Interestingly, the diabetogenic effect of GSK690693 is not reverted by either metformin or pioglitazone or GLP-I agonist exendin-4, but only by a low-carbohydrate diet (Rhodes et al. 2008, Crouthamel et al. 2009).
A further class of targeted drugs that may interfere with blood glucose levels is the inhibitors of the mTOR kinase. This mTOR serine/threonine kinase and the mTOR-raptor complex (TORC1) regulate cell cycle progression (i.e. G1-S phase transition) and increase the expression of angiogenic factors. When dysregulated, mTOR plays a key role in cell proliferation and neoplastic transformation favoring the development of resistance to several types of cancer therapy (Bjornsti & Houghton 2004, Panwalkar et al. 2004).
Several mTOR inhibitors have been developed in vitro (Fig. 5). Some of them have been used in clinical trials. The most important, everolimus (RAD001; 40-O-2-hydroxyethyl-rapamycil), an orally available ester derivative of the anti-fungal antibiotic sirolimus (rapamycin), is currently used as an immunosuppressive agent to prevent rejection in transplant recipients (Eisen et al. 2003, Lorber et al. 2005). Immunosuppression maintenance with everolimus has been associated with a significantly reduced risk of developing de novo malignancies after renal transplant (Kauffman et al. 2005). Everolimus forms a complex with the immunophilin FKBP-12, which then binds to and disrupts TORC1, leading to mTOR inhibition and G1 phase cell cycle arrest, apoptosis (Aguirre et al. 2004, Majumder et al. 2004), and angiogenesis suppression (Majumder et al. 2004). Temsirolimus is a further novel mTOR inhibitor of the same family, recently approved for the treatment of renal cell carcinoma with unfavorable clinical characteristics. As expected from mTOR inhibition, hyperglycemia, hypertriglyceridemia, and hypercholesterolemia have been observed in 20% of patients treated with these inhibitors. In particular, recent data have reported increased blood glucose levels in 26% of temsirolimus-treated patients with 11% displaying G3/G4 hyperglycemia (Bellmunt et al. 2008, Malizzia & Hsu 2008). Most diabetic patients treated with temsirolimus required an increase in their hypoglycemic treatment, and roughly 30% of nondiabetic patients had to begin a specific therapy to lower their blood glucose (Bellmunt et al. 2008, Malizzia & Hsu 2008). Treatment of mTOR inhibition-related hyperglycemia has not yet been studied.
Finally, inhibitors of the ABL TK may also affect glucose homeostasis. In vitro results indicate that ABL is involved in IR signaling and upon insulin stimulation enhances IR-dependent metabolic effects while attenuating the nonmetabolic ones (Frasca et al. 2007, Genua et al. 2009). Therefore, treatment with an ABL inhibitor was expected to impair glucose homeostasis (Fig. 6). However, adult patients with chronic and accelerated phase chronic myelogenous leukemia (CML), treated with the ABL inhibitor imatinib mesylate, have actually shown a consistent reduction in their blood glucose levels (Veneri et al. 2005). Interestingly, a recent report has described hyperglycemia in 10% of CML patients treated with nilotinib, a second-generation ABL kinase inhibitor currently used for individuals resistant or intolerant to imatinib (Kantarjian et al. 2006). The increase in fasting glucose registered after nilotinib therapy is predictive of drug response and apparently does not require administration of hypoglycemic drugs (Deremer et al. 2008). However, the follow-up of the study is too short to yield conclusive evidence, especially considering that patients responding to nilotinib will have to continue drug treatment indefinitely until disease progression.
|
| Conclusions |
|---|
Because of the intrinsic heterogeneity of both diabetes and cancer, studies on the association of the two diseases are not easy to carry out. Indeed, considering the wide array of possible mechanisms causing increased cancer incidence and mortality in diabetic patients, it is difficult to accurately define the aims, the recruitment criteria, and the appropriate design for such studies.
The available evidence indicates that the level of cancer risk related to diabetes will probably differ for each diabetic patient, on the basis of the cancer type and many other diabetes-related factors. Our present knowledge provides good evidence for a mild increase of cancer risk (and cancer mortality) in diabetic patients, more evident for some site-specific cancers. Present evidence, however, does not allow us to accurately define the general and the specific organ cancer risks in the individual diabetic patient. Because of the growing worldwide frequency of diabetes, this question needs to be properly addressed, in order to acquire a more rational approach to cancer prevention and treatment in diabetic patients.
| Declaration of interest |
|---|
| Funding |
|---|
| Acknowledgements |
|---|
| References |
|---|
Aguirre D, Boya P, Bellet D, Faivre S, Troalen F, Benard J, Saulnier P, Hopkins-Donaldson S, Zangemeister-Wittke U, Kroemer G et al. 2004 Bcl-2 and CCND1/CDK4 expression levels predict the cellular effects of mTOR inhibitors in human ovarian carcinoma. Apoptosis 9 797-805.[CrossRef][Web of Science][Medline]
Aiello A, Pandini G, Frasca F, Conte E, Murabito A, Sacco A, Genua M, Vigneri R & Belfiore A 2006 Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology 147 4463-4475.
Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE & Thor AD 2009 Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 8 909-915.[Web of Science][Medline]
Barone BB, Yeh HC, Snyder CF, Peairs KS, Stein KB, Derr RL, Wolff AC & Brancati FL 2008 Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. Journal of the American Medical Association 300 2754-2764.
Barrett-Connor E 1992 Lower endogenous androgen levels and dyslipidemia in men with non-insulin-dependent diabetes mellitus. Annals of Internal Medicine 117 807-811.
Basso D, Valerio A, Seraglia R, Mazza S, Piva MG, Greco E, Fogar P, Gallo N, Pedrazzoli S, Tiengo A et al. 2002 Putative pancreatic cancer-associated diabetogenic factor: 2030 MW peptide. Pancreas 24 8-14.[CrossRef][Web of Science][Medline]
Bellmunt J, Szczylik C, Feingold J, Strahs A & Berkenblit A 2008 Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Annals of Oncology 19 1387-1392.
Betancourt-Albrecht M & Cunningham GR 2003 Hypogonadism and diabetes. International Journal of Impotence Research 15 S14-S20.[CrossRef][Web of Science][Medline]
Bjornsti MA & Houghton PJ 2004 The TOR pathway: a target for cancer therapy. Nature Reviews. Cancer 4 335-348.[CrossRef][Web of Science][Medline]
Bonovas S, Filioussi K & Tsantes A 2004 Diabetes mellitus and risk of prostate cancer: a meta-analysis. Diabetologia 47 1071-1078.[Web of Science][Medline]
Bowker SL, Majumdar SR, Veugelers P & Johnson JA 2006 Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 29 254-258.
Buzzetti R, Di Pietro S, Giaccari A, Petrone A, Locatelli M, Suraci C, Capizzi M, Arpi ML, Bazzigaluppi E, Dotta F et al. 2007 High titer of autoantibodies to GAD identifies a specific phenotype of adult-onset autoimmune diabetes. Diabetes Care 30 932-938.
Caldwell S & Lazo M 2009 Is exercise an effective treatment for NASH? Knowns and unknowns. Annals of Hepatology 8 S60-S66.
Calle EE, Rodriguez C, Walker-Thurmond K & Thun MJ 2003 Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. New England Journal of Medicine 348 1625-1638.
Cazzaniga M, Bonanni B, Guerrieri-Gonzaga A & Decensi A 2009 Is it time to test metformin in breast cancer clinical trials? Cancer Epidemiology, Biomarkers and Prevention 18 701-705.
Cebioglu M, Schild HH & Golubnitschaja O 2008 Diabetes mellitus as a risk factor for cancer: stress or viral etiology? Infectious Disorders - Drug Targets 8 76-87.[Medline]
Chari ST, Leibson CL, Rabe KG, Ransom J, de Andrade M & Petersen GM 2005 Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 129 504-511.[Web of Science][Medline]
Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, de Andrade M & Petersen GM 2008 Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 134 95-101.[CrossRef][Web of Science][Medline]
Chen HF, Li CY, Chen P, See TT & Lee HY 2006 Seroprevalence of hepatitis B and C in type 2 diabetic patients. Journal of the Chinese Medical Association 69 146-152.[CrossRef][Medline]
Chow WH, Gridley G, Fraumeni JF Jr & Jarvholm B 2000 Obesity, hypertension, and the risk of kidney cancer in men. New England Journal of Medicine 343 1305-1311.
Claycombe KJ, Jones BH, Standridge MK, Guo Y, Chun JT, Taylor JW & Moustaid-Moussa N 1998 Insulin increases fatty acid synthase gene transcription in human adipocytes. American Journal of Physiology 274 R1253-R1259.[Web of Science][Medline]
Cleary MP & Grossmann ME 2009 Minireview: obesity and breast cancer: the estrogen connection. Endocrinology 150 2537-2542.
Cleary MP, Ray A, Rogozina OP, Dogan S & Grossmann ME 2009 Targeting the adiponectin:leptin ratio for postmenopausal breast cancer prevention. Frontiers in Bioscience 1 329-357.
Corbould A, Zhao H, Mirzoeva S, Aird F & Dunaif A 2006 Enhanced mitogenic signaling in skeletal muscle of women with polycystic ovary syndrome. Diabetes 55 751-759.
Coughlin SS, Calle EE, Teras LR, Petrelli J & Thun MJ 2004 Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. American Journal of Epidemiology 159 1160-1167.
Crouthamel MC, Kahana JA, Korenchuk S, Zhang SY, Sundaresan G, Eberwein DJ, Brown KK & Kumar R 2009 Mechanism and management of AKT inhibitor-induced hyperglycemia. Clinical Cancer Research 15 217-225.
Dankner R, Chetrit A & Segal P 2007 Glucose tolerance status and 20 year cancer incidence. Israel Medical Association Journal 9 592-596.[Web of Science][Medline]
Davila JA, Morgan RO, Shaib Y, McGlynn KA & El-Serag HB 2005 Diabetes increases the risk of hepatocellular carcinoma in the United States: a population based case-control study. Gut 54 533-539.
De Block CE, De Leeuw IH & Van Gaal LF 1999 High prevalence of manifestations of gastric autoimmunity in parietal cell antibody-positive type 1 (insulin-dependent) diabetic patients. The Belgian Diabetes Registry. Journal of Clinical Endocrinology and Metabolism 84 4062-4067.
De Meyts P, Christoffersen CT, Urso B, Wallach B, Gronskov K, Yakushiji F & Shymko RM 1995 Role of the time factor in signaling specificity: application to mitogenic and metabolic signaling by the insulin and insulin-like growth factor-I receptor tyrosine kinases. Metabolism 44 2-11.[CrossRef][Web of Science][Medline]
Deremer DL, Ustun C & Natarajan K 2008 Nilotinib: a second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clinical Therapeutics 30 1956-1975.[CrossRef][Web of Science][Medline]
Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sorensen K, Hummel M, Lind JM et al. 2003 Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients. New England Journal of Medicine 349 847-858.
El-Serag HB, Hampel H & Javadi F 2006 The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clinical Gastroenterology and Hepatology 4 369-380.[CrossRef][Web of Science]
Elwing JE, Gao F, Davidson NO & Early DS 2006 Type 2 diabetes mellitus: the impact on colorectal adenoma risk in women. American Journal of Gastroenterology 101 1866-1871.[CrossRef][Web of Science][Medline]
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR & Morris AD 2005 Metformin and reduced risk of cancer in diabetic patients. BMJ 330 1304-1305.
Everhart J & Wright D 1995 Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. Journal of the American Medical Association 273 1605-1609.
Federico A, Morgillo F, Tuccillo C, Ciardiello F & Loguercio C 2007 Chronic inflammation and oxidative stress in human carcinogenesis. International Journal of Cancer 121 2381-2386.[CrossRef][Web of Science][Medline]
Fernandez AM, Kim JK, Yakar S, Dupont J, Hernandez-Sanchez C, Castle AL, Filmore J, Shulman GI & Le Roith D 2001 Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes and Development 15 1926-1934.
Ferrannini E & Cobelli C 1987 The kinetics of insulin in man. II. Role of the liver. Diabetes/Metabolism Reviews 3 365-397.[Medline]
Folsom AR, Anderson KE, Sweeney C & Jacobs DR Jr 2004 Diabetes as a risk factor for death following endometrial cancer. Gynecologic Oncology 94 740-745.[CrossRef][Web of Science][Medline]
Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A & Vigneri R 1999 Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Molecular and Cellular Biology 19 3278-3288.
Frasca F, Pandini G, Malaguarnera R, Mandarino A, Messina RL, Sciacca L, Belfiore A & Vigneri R 2007 Role of c-Abl in directing metabolic versus mitogenic effects in insulin receptor signaling. Journal of Biological Chemistry 282 26077-26088.
Friberg E, Orsini N, Mantzoros CS & Wolk A 2007 Diabetes mellitus and risk of endometrial cancer: a meta-analysis. Diabetologia 50 1365-1374.[CrossRef][Web of Science][Medline]
Gable KL, Maddux BA, Penaranda C, Zavodovskaya M, Campbell MJ, Lobo M, Robinson L, Schow S, Kerner JA, Goldfine ID et al. 2006 Diarylureas are small-molecule inhibitors of insulin-like growth factor I receptor signaling and breast cancer cell growth. Molecular Cancer Therapeutics 5 1079-1086.
Gapstur SM, Gann PH, Lowe W, Liu K, Colangelo L & Dyer A 2000 Abnormal glucose metabolism and pancreatic cancer mortality. Journal of the American Medical Association 283 2552-2558.
Genua M, Pandini G, Cassarino MF, Messina RL & Frasca F 2009 Chapter 4 c-Abl and insulin receptor signalling. Vitamins and Hormones 80 77-105.[CrossRef][Web of Science][Medline]
Glass AG, Lacey JV Jr, Carreon JD & Hoover RN 2007 Breast cancer incidence, 1980-2006: combined roles of menopausal hormone therapy, screening mammography, and estrogen receptor status. Journal of the National Cancer Institute 99 1152-1161.
Goodwin PJ, Ennis M, Fantus IG, Pritchard KI, Trudeau ME, Koo J & Hood N 2005 Is leptin a mediator of adverse prognostic effects of obesity in breast cancer? Journal of Clinical Oncology 23 6037-6042.
Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, Kim PJ, Owens RJ & Lang NP 2007 Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. Journal of Clinical Oncology 25 1476-1481.
Green A & Jensen OM 1985 Frequency of cancer among insulin-treated diabetic patients in Denmark. Diabetologia 28 128-130.[Web of Science][Medline]
Haluska P, Carboni JM, Loegering DA, Lee FY, Wittman M, Saulnier MG, Frennesson DB, Kalli KR, Conover CA, Attar RM et al. 2006 In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Research 66 362-371.
Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, Yap TA, Roberts ML, Sharma A, Gualberto A et al. 2007 Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clinical Cancer Research 13 5834-5840.
Harris MI, Flegal KM, Cowie CC, Eberhardt MS, Goldstein DE, Little RR, Wiedmeyer HM & Byrd-Holt DD 1998 Prevalence of diabetes, impaired fasting glucose, and impaired glucose tolerance in U.S. adults. The Third National Health and Nutrition Examination Survey, 1988-1994. Diabetes Care 21 518-524.[Abstract]
Heuson JC & Legros N 1972 Influence of insulin deprivation on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats subjected to alloxan diabetes and food restriction. Cancer Research 32 226-232.
Heuson JC, Legros N & Heimann R 1972 Influence of insulin administration on growth of the 7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in intact, oophorectomized, and hypophysectomized rats. Cancer Research 32 233-238.
Hillgartner FB, Salati LM & Goodridge AG 1995 Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiological Reviews 75 47-76.
Hofmann F & Garcia-Echeverria C 2005 Blocking the insulin-like growth factor-I receptor as a strategy for targeting cancer. Drug Discovery Today 10 1041-1047.[CrossRef][Web of Science][Medline]
Hu X, Juneja SC, Maihle NJ & Cleary MP 2002 Leptin - a growth factor in normal and malignant breast cells and for normal mammary gland development. Journal of the National Cancer Institute 94 1704-1711.
Hudgins LC 2000 Effect of high-carbohydrate feeding on triglyceride and saturated fatty acid synthesis. Proceedings of the Society for Experimental Biology and Medicine 225 178-183.
Hudgins LC, Hellerstein MK, Seidman CE, Neese RA, Tremaroli JD & Hirsch J 2000 Relationship between carbohydrate-induced hypertriglyceridemia and fatty acid synthesis in lean and obese subjects. Journal of Lipid Research 41 595-604.
Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A, Barzi F & Woodward M 2005 Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. British Journal of Cancer 92 2076-2083.[CrossRef][Web of Science][Medline]
Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R et al. 2004 Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Molecular Cancer Therapeutics 3 763-772.
Ihle NT, Lemos R Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL & Powis G 2009a Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Research 69 143-150.
Ihle NT, Lemos R, Schwartz D, Oh J, Halter RJ, Wipf P, Kirkpatrick L & Powis G 2009b Peroxisome proliferator-activated receptor gamma agonist pioglitazone prevents the hyperglycemia caused by phosphatidylinositol 3-kinase pathway inhibition by PX-866 without affecting antitumor activity. Molecular Cancer Therapeutics 8 94-100.
Ji QS, Mulvihill MJ, Rosenfeld-Franklin M, Cooke A, Feng L, Mak G, O'Connor M, Yao Y, Pirritt C, Buck E et al. 2007 A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Molecular Cancer Therapeutics 6 2158-2167.
Kaaks R 1996 Nutrition, hormones, and breast cancer: is insulin the missing link? Cancer Causes and Control 7 605-625.[CrossRef][Web of Science][Medline]
Kaaks R & Lukanova A 2001 Energy balance and cancer: the role of insulin and insulin-like growth factor-I. Proceedings of the Nutrition Society 60 91-106.[Web of Science][Medline]
Kalli KR, Falowo OI, Bale LK, Zschunke MA, Roche PC & Conover CA 2002 Functional insulin receptors on human epithelial ovarian carcinoma cells: implications for IGF-II mitogenic signaling. Endocrinology 143 3259-3267.
Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W et al. 2006 Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. New England Journal of Medicine 354 2542-2551.
Kasper JS & Giovannucci E 2006 A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiology, Biomarkers and Prevention 15 2056-2062.
Kauffman HM, Cherikh WS, Cheng Y, Hanto DW & Kahan BD 2005 Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation 80 883-889.[CrossRef][Web of Science][Medline]
Keating NL, O'Malley AJ & Smith MR 2006 Diabetes and cardiovascular disease during androgen deprivation therapy for prostate cancer. Journal of Clinical Oncology 24 4448-4456.
Kern PA, Ranganathan S, Li C, Wood L & Ranganathan G 2001 Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. American Journal of Physiology. Endocrinology and Metabolism 280 E745-E751.
Key TJ, Appleby PN, Reeves GK, Roddam A, Dorgan JF, Longcope C, Stanczyk FZ, Stephenson HE Jr, Falk RT, Miller R et al. 2003 Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. Journal of the National Cancer Institute 95 1218-1226.
Koro C, Barrett S & Qizilbash N 2007 Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiology and Drug Safety 16 485-492.[CrossRef][Web of Science][Medline]
von Kriegstein E & von Kriegstein K 2007 Inhaled insulin for diabetes mellitus. New England Journal of Medicine 356 2106.
Krone CA & Ely JT 2005 Controlling hyperglycemia as an adjunct to cancer therapy. Integrated Cancer Therapy 4 25-31.[CrossRef]
Kuzuya H, Matsuura N, Sakamoto M, Makino H, Sakamoto Y, Kadowaki T, Suzuki Y, Kobayashi M, Akazawa Y, Nomura M et al. 1993 Trial of insulinlike growth factor I therapy for patients with extreme insulin resistance syndromes. Diabetes 42 696-705.[Abstract]
Lacy MQ, Alsina M, Fonseca R, Paccagnella ML, Melvin CL, Yin D, Sharma A, Enriquez Sarano M, Pollak M, Jagannath S et al. 2008 Phase I, pharmacokinetic and pharmacodynamic study of the anti-insulinlike growth factor type 1 receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. Journal of Clinical Oncology 26 3196-3203.
Larsson SC, Orsini N & Wolk A 2005 Diabetes mellitus and risk of colorectal cancer: a meta-analysis. Journal of the National Cancer Institute 97 1679-1687.
Larsson SC, Orsini N, Brismar K & Wolk A 2006 Diabetes mellitus and risk of bladder cancer: a meta-analysis. Diabetologia 49 2819-2823.[CrossRef][Web of Science][Medline]
Larsson SC, Mantzoros CS & Wolk A 2007 Diabetes mellitus and risk of breast cancer: a meta-analysis. International Journal of Cancer 121 856-862.[CrossRef][Web of Science][Medline]
Limburg PJ, Vierkant RA, Fredericksen ZS, Leibson CL, Rizza RA, Gupta AK, Ahlquist DA, Melton LJ III, Sellers TA & Cerhan JR 2006 Clinically confirmed type 2 diabetes mellitus and colorectal cancer risk: a population-based, retrospective cohort study. American Journal of Gastroenterology 101 1872-1879.[CrossRef][Web of Science][Medline]
Lindblad P & Adami HO 2002 Kidney cancer. In Textbook of Cancer Epidemiology, pp 467-485 Eds D Hunter, HO Adami & D Trichopoulos. New York: Oxford University Press.
Lindblad P, Chow WH, Chan J, Bergstrom A, Wolk A, Gridley G, McLaughlin JK, Nyren O & Adami HO 1999 The role of diabetes mellitus in the aetiology of renal cell cancer. Diabetologia 42 107-112.[CrossRef][Web of Science][Medline]
Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR & Hux JE 2008 The impact of diabetes on survival following breast cancer. Breast Cancer Research and Treatment 109 389-395.[CrossRef][Web of Science][Medline]
LoPiccolo J, Blumenthal GM, Bernstein WB & Dennis PA 2008 Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resistance Updates 11 32-50.[CrossRef][Web of Science][Medline]
Lorber MI, Mulgaonkar S, Butt KM, Elkhammas E, Mendez R, Rajagopalan PR, Kahan B, Sollinger H, Li Y, Cretin N et al. 2005 Everolimus versus mycophenolate mofetil in the prevention of rejection in de novo renal transplant recipients: a 3-year randomized, multicenter, phase III study. Transplantation 80 244-252.[CrossRef][Web of Science][Medline]
Luo Z, Saha AK, Xiang X & Ruderman NB 2005 AMPK, the metabolic syndrome and cancer. Trends in Pharmacological Sciences 26 69-76.[CrossRef][Medline]
Lupu R & Menendez JA 2006a Pharmacological inhibitors of fatty acid synthase (FASN) - catalyzed endogenous fatty acid biogenesis: a new family of anti-cancer agents? Current Pharmaceutical Biotechnology 7 483-493.[CrossRef][Web of Science][Medline]
Lupu R & Menendez JA 2006b Targeting fatty acid synthase in breast and endometrial cancer: an alternative to selective estrogen receptor modulators? Endocrinology 147 4056-4066.[CrossRef][Web of Science][Medline]
Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, Gaziano JM, Pollak M & Stampfer MJ 2008 Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncology 9 1039-1047.[CrossRef][Web of Science][Medline]
Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR et al. 2004 mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nature Medicine 10 594-601.[CrossRef][Web of Science][Medline]
Malizzia LJ & Hsu A 2008 Temsirolimus, an mTOR inhibitor for treatment of patients with advanced renal cell carcinoma. Clinical Journal of Oncology Nursing 12 639-646.[CrossRef][Web of Science][Medline]
McCarty MF 2004 Chronic activation of AMP-activated kinase as a strategy for slowing aging. Medical Hypotheses 63 334-339.[CrossRef][Web of Science][Medline]
Menendez JA, Vazquez-Martin A, Ortega FJ & Fernandez-Real JM 2009 Fatty acid synthase: association with insulin resistance, type 2 diabetes, and cancer. Clinical Chemistry 55 425-438.
Meyerhardt JA, Catalano PJ, Haller DG, Mayer RJ, Macdonald JS, Benson AB III & Fuchs CS 2003 Impact of diabetes mellitus on outcomes in patients with colon cancer. Journal of Clinical Oncology 21 433-440.
Mitri J & Pittas AG 2009 Inhaled insulin - what went wrong. Nature Clinical Practice. Endocrinology and Metabolism 5 24-25.
Mitri J, Castillo J & Pittas AG 2008 Diabetes and risk of non-Hodgkin's lymphoma: a meta-analysis of observational studies. Diabetes Care 31 2391-2397.
Moller DE, Yokota A, Caro JF & Flier JS 1989 Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Molecular Endocrinology 3 1263-1269.
Monami M, Balzi D, Lamanna C, Barchielli A, Masotti G, Buiatti E, Marchionni N & Mannucci E 2007 Are sulphonylureas all the same? A cohort study on cardiovascular and cancer-related mortality. Diabetes/Metabolism Research and Reviews 23 479-484.[CrossRef]
Moustaid N, Jones BH & Taylor JW 1996 Insulin increases lipogenic enzyme activity in human adipocytes in primary culture. Journal of Nutrition 126 865-870.
Mulvihill MJ, Ji QS, Coate HR, Cooke A, Dong H, Feng L, Foreman K, Rosenfeld-Franklin M, Honda A, Mak G et al. 2008 Novel 2-phenylquinolin-7-yl-derived imidazo[1,5-a]pyrazines as potent insulin-like growth factor-I receptor (IGF-IR) inhibitors. Bioorganic and Medicinal Chemistry 16 1359-1375.[CrossRef][Medline]
Noy A & Bilezikian JP 1994 Clinical review 63: diabetes and pancreatic cancer: clues to the early diagnosis of pancreatic malignancy. Journal of Clinical Endocrinology and Metabolism 79 1223-1231.[Abstract]
Ohshima H, Tatemichi M & Sawa T 2003 Chemical basis of inflammation-induced carcinogenesis. Archives of Biochemistry and Biophysics 417 3-11.[CrossRef][Web of Science][Medline]
Oldenburg B, Diepersloot RJ & Hoekstra JB 1996 High seroprevalence of Helicobacter pylori in diabetes mellitus patients. Digestive Diseases and Sciences 41 458-461.[CrossRef][Web of Science][Medline]
Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM & Chari ST 2008 Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology 134 981-987.[CrossRef][Web of Science][Medline]
Pannala R, Basu A, Petersen GM & Chari ST 2009 New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncology 10 88-95.[CrossRef][Web of Science][Medline]
Panwalkar A, Verstovsek S & Giles FJ 2004 Mammalian target of rapamycin inhibition as therapy for hematologic malignancies. Cancer 100 657-666.[CrossRef][Web of Science][Medline]
Papa V, Pezzino V, Costantino A, Belfiore A, Giuffrida D, Frittitta L, Vannelli GB, Brand R, Goldfine ID & Vigneri R 1990 Elevated insulin receptor content in human breast cancer. Journal of Clinical Investigation 86 1503-1510.[Web of Science][Medline]
Pennisi P, Gavrilova O, Setser-Portas J, Jou W, Santopietro S, Clemmons D, Yakar S & LeRoith D 2006 Recombinant human insulin-like growth factor-I treatment inhibits gluconeogenesis in a transgenic mouse model of type 2 diabetes mellitus. Endocrinology 147 2619-2630.
Pollak M 2008 Insulin and insulin-like growth factor signalling in neoplasia. Nature Reviews. Cancer 8 915-928.[CrossRef][Web of Science][Medline]
Postic C & Girard J 2008 The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes and Metabolism 34 643-648.[CrossRef]
Ramos-Nino ME, MacLean CD & Littenberg B 2007 Association between cancer prevalence and use of thiazolidinediones: results from the Vermont Diabetes Information System. BMC Medicine 5 17.[CrossRef][Medline]
Reed MJ & Purohit A 2001 Aromatase regulation and breast cancer. Clinical Endocrinology 54 563-571.[CrossRef][Medline]
Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, McConnell RT, Gilmer TM, Zhang SY, Robell K et al. 2008 Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Research 68 2366-2374.
del Rincon JP, Iida K, Gaylinn BD, McCurdy CE, Leitner JW, Barbour LA, Kopchick JJ, Friedman JE, Draznin B & Thorner MO 2007 Growth hormone regulation of p85alpha expression and phosphoinositide 3-kinase activity in adipose tissue: mechanism for growth hormone-mediated insulin resistance. Diabetes 56 1638-1646.[CrossRef][Web of Science][Medline]
Ruderman N & Prentki M 2004 AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nature Reviews. Drug Discovery 3 340-351.[CrossRef][Web of Science][Medline]
Saylor PJ & Smith MR 2009 Metabolic complications of androgen deprivation therapy for prostate cancer. Journal of Urology 181 1998-2006.[CrossRef][Web of Science][Medline]
Sciacca L, Costantino A, Pandini G, Mineo R, Frasca F, Scalia P, Sbraccia P, Goldfine ID, Vigneri R & Belfiore A 1999 Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene 18 2471-2479.[CrossRef][Web of Science][Medline]
Semenkovich CF, Coleman T & Fiedorek FT Jr 1995 Human fatty acid synthase mRNA: tissue distribution, genetic mapping, and kinetics of decay after glucose deprivation. Journal of Lipid Research 36 1507-1521.[Abstract]
Stadler J, Yeung KS, Furrer R, Marcon N, Himal HS & Bruce WR 1988 Proliferative activity of rectal mucosa and soluble fecal bile acids in patients with normal colons and in patients with colonic polyps or cancer. Cancer Letters 38 315-320.[CrossRef][Web of Science][Medline]
Stevens RJ, Roddam AW & Beral V 2007 Pancreatic cancer in type 1 and young-onset diabetes: systematic review and meta-analysis. British Journal of Cancer 96 507-509.[CrossRef][Web of Science][Medline]
Szlosarek P, Charles KA & Balkwill FR 2006 Tumour necrosis factor-alpha as a tumour promoter. European Journal of Cancer 42 745-750.[CrossRef][Web of Science][Medline]
Turturro F, Friday E & Welbourne T 2007 Hyperglycemia regulates thioredoxin-ROS activity through induction of thioredoxin-interacting protein (TXNIP) in metastatic breast cancer-derived cells MDA-MB-231. BMC Cancer 7 96.[CrossRef][Medline]
Vasilcanu R, Vasilcanu D, Rosengren L, Natalishvili N, Sehat B, Yin S, Girnita A, Axelson M, Girnita L & Larsson O 2008 Picropodophyllin induces downregulation of the insulin-like growth factor 1 receptor: potential mechanistic involvement of Mdm2 and β-arrestin1. Oncogene 27 1629-1638.[CrossRef][Web of Science][Medline]
Vella V, Pandini G, Sciacca L, Mineo R, Vigneri R, Pezzino V & Belfiore A 2002 A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. Journal of Clinical Endocrinology and Metabolism 87 245-254.
Veneri D, Franchini M & Bonora E 2005 Imatinib and regression of type 2 diabetes. New England Journal of Medicine 352 1049-1050.
Vigneri P, Frasca F, Sciacca L, Frittitta L & Vigneri R 2006 Obesity and cancer. Nutrition, Metabolism, and Cardiovascular Diseases 16 1-7.[CrossRef][Web of Science][Medline]
Washio M, Mori M, Khan M, Sakauchi F, Watanabe Y, Ozasa K, Hayashi K, Miki T, Nakao M, Mikami K et al. 2007 Diabetes mellitus and kidney cancer risk: the results of Japan Collaborative Cohort Study for Evaluation of Cancer Risk (JACC Study). International Journal of Urology 14 393-397.[CrossRef][Web of Science][Medline]
Will JC, Galuska DA, Vinicor F & Calle EE 1998 Colorectal cancer: another complication of diabetes mellitus? American Journal of Epidemiology 147 816-825.
Williams JA & Goldfine ID 1985 The insulin-pancreatic acinar axis. Diabetes 34 980-986.[Abstract]
Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, Bouxsein ML, Kopchick JJ & LeRoith D 2004 Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. Journal of Clinical Investigation 113 96-105.[CrossRef][Web of Science][Medline]
Yuan JM, Castelao JE, Gago-Dominguez M, Ross RK & Yu MC 1998 Hypertension, obesity and their medications in relation to renal cell carcinoma. British Journal of Cancer 77 1508-1513.[Web of Science][Medline]
Zendehdel K, Nyren O, Ostenson CG, Adami HO, Ekbom A & Ye W 2003 Cancer incidence in patients with type 1 diabetes mellitus: a population-based cohort study in Sweden. Journal of the National Cancer Institute 95 1797-1800.
Zimmermann K, Wittman MD, Saulnier MG, Velaparthi U, Langley DR, Sang X, Frennesson D, Carboni J, Li A, Greer A et al. 2008 Balancing oral exposure with Cyp3A4 inhibition in benzimidazole-based IGF-IR inhibitors. Bioorganic and Medicinal Chemistry Letters 18 4075-4080.[CrossRef]
Zucchetto A, Dal Maso L, Tavani A, Montella M, Ramazzotti V, Talamini R, Canzonieri V, Garbeglio A, Negri E, Franceschi S et al. 2007 History of treated hypertension and diabetes mellitus and risk of renal cell cancer. Annals of Oncology 18 596-600.
This article has been cited by other articles:
 |
E. Giovannucci, D. M. Harlan, M. C. Archer, R. M. Bergenstal, S. M. Gapstur, L. A. Habel, M. Pollak, J. G. Regensteiner, and D. Yee Diabetes and Cancer: A consensus report Diabetes Care, July 1, 2010; 33(7): 1674 - 1685. [Full Text] [PDF] |
||||
|
|
K. Hemminki, X. Li, J. Sundquist, and K. Sundquist Risk of Cancer Following Hospitalization for Type 2 Diabetes Oncologist, June 1, 2010; 15(6): 548 - 555. [Abstract] [Full Text] [PDF] |
||||
|
|
H. C. Gerstein Does Insulin Therapy Promote, Reduce, or Have a Neutral Effect on Cancers? JAMA, February 3, 2010; 303(5): 446 - 447. [Full Text] [PDF] |
||||
|
|
C. R. Chong and B. A. Chabner Mysterious Metformin Oncologist, December 1, 2009; 14(12): 1178 - 1181. [Full Text] [PDF] |
||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |