The pleiotropic roles of transforming growth factor beta in homeostasis and carcinogenesis of endocrine organs
- Life Sciences Division, 1 Cyclotron Road, MS 977-225A, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
- (Requests for offprints should be addressed to M H Barcellos-Hoff; Email: MHBarcellos-Hoff{at}lbl.gov)
Abstract
Transforming growth factor β (TGF-β) is a ubiquitous cytokine that plays a critical role in numerous pathways regulating cellular and tissue homeostasis. TGF-β is regulated by hormones and is a primary mediator of hormone response in uterus, prostate and mammary glands. This review will address the role of TGF-β in regulating hormone-dependent proliferation and morphogenesis. The subversion of TGF-β regulation during the processes of carcinogenesis, with particular emphasis on its effects on genetic stability and epithelial to mesenchymal transition, will also be examined. An understanding of the multiple and complex mechanisms of TGF-β regulation of epithelial function, and the ultimate loss of TGF-β function during carcinogenesis, will be critical in the design of novel therapeutic interventions for endocrine-related cancers.
Introduction
The members of the TGF-β family are highly conserved in mammals and are involved in a variety of cellular functions from embryo development to adult tissue homeostasis (Herpin et al. 2004). TGF-β1, which is the founding member and often referred to as the ‘prototype’ of this family, was originally named based on its ability to stimulate anchorage independent growth in fibroblasts (Roberts et al. 1981). However, in many respects, transforming is a misnomer since in addition to this transforming capacity, TGF-β stringently regulates epithelial growth control, mediates DNA damage repair, and stimulates apoptosis and stem cell function (Fig. 1⇓). Thus, TGF-β demonstrates both oncogenic and tumor suppressive properties (reviewed in Derynck et al. 2001). This review will highlight the functions of TGF-β on hormone-dependent epithelia ranging from the regulation of normal development and homeostasis to its role in oncogenesis and progression. Our studies using in vitro and in vivo models of mammary development and radiation-induced carcinogenesis will be used to illustrate several TGF-β roles.
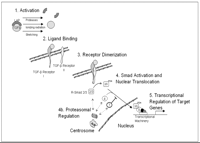
Canonical TGF-β signaling pathway. As many cells express TGF-β and its receptors, the restriction of TGF-β activation is a key regulator of its activity. TGF-β activation occurs through multiple mechanisms including proteases, heat, ionizing radiation and mechanical stretching. Once activated, TGF-β binds to TGF-β receptor I leading to dimerization with TGF-β receptor II and activation of receptor Smad (R-Smad) complexes. Upon activation, R-Smads translocate to the nucleus and complex with the cellular transcriptional machinery to regulate gene expression. R-Smad activation and translocation is subject to proteasomal regulation and additional regulation by inhibitory-Smad complexes, including Smad7.
TGF-β activation
There are three mammalian Tgfβ genes (Tgfβ1, Tgfβ2, and Tgfβ3), for which there is a high degree of sequence homology and some functional overlap. This review will focus on TGF-β1. Nearly all cells secrete latent TGF-β and express the TGF-β receptors making the restriction of TGF-β activation a key regulator for its biological effects. Recent detailed reviews on TGF-β activation and signaling are available (Yue & Mulder 2001, Siegel & Massague 2003, Rifkin 2005).
TGF-β can act in an autocrine, paracrine or endocrine fashion (Smith 1996). All TGF-β isoforms are secreted as inactive, or latent, forms and upon activation can potentially bind three surface receptors (TβR I–III). The ubiquitously secreted and inactive form is called ‘latent complex (LTGF-β)’ and it consists of the mature TGF-β dimer non-covalently bound to its latency associated peptide (LAP). In addition, some LTGF-β is bound to latent TGF-β binding protein (LTBP) (Rifkin 2005). Most cells, including epithelial, stromal, and immune cells such as macrophages, make TGF-β and have receptors for the ligand. The secretion of TGF-β in latent forms is a principle regulatory event that restricts its biological availability and makes LTGF-β activation the key to understanding its activity in situ.
In addition to its regulatory functions, LTBP provides a means of anchoring latent TGF-β within the extracellular matrix (ECM) (Barcellos-Hoff & Ewan 2000). Fibronectin (Fn) provides an initial scaffold that precedes and patterns LTBP-1 deposition (Dallas et al. 2005). Fn is also required for the continued assembly of LTBP1 into the ECM of osteoblasts and fibroblasts (Dallas et al. 2005). Cleavage of the LAP and the adjacent LTBP, by several proteases such as plasmin, thrombin, plasma transglutaminases and endoglycosylases, is an initial step in the activation of TGF-β (Javelaud & Mauviel 2004). Recent evidence demonstrates that LTBP-1 enables αvβ6 integrin-mediated activation by both fixing and concentrating the latent complex in the ECM leading to a mechanical stretching of the LTGF-β molecule (Annes et al. 2004). In addition to the aforementioned mechanisms, LTGF-β is efficiently activated by exposure to reactive oxygen species that may be generated by ionizing radiation and other sources (Barcellos-Hoff et al. 1994, Barcellos-Hoff 1996).
Using antibodies specific for latent and active TGF-β we were able to show that TGF-β activation in the mouse mammary gland is an event that is spatially and temporally highly restricted (Ewan et al. 2002a). Active TGF-β localized predominantly to the luminal epithelium and is undetectable in myoepithelial cells and is weakly detected in normal stroma. Within the luminal epithelium during phases of proliferation (puberty, estrus or pregnancy), active TGF-β1 is restricted to certain cells. Alterations in immunolocalization pattern, from hetero- to homogeneous, as a function of the estrous cycle suggested that TGF-β activation in the mammary gland is regulated by ovarian hormones.
TGF-β signaling
Once activation releases TGF-β from the latent complex, it can bind to ubiquitously expressed surface receptors of target cells (Fig. 1⇑). The type I (TβRI) and type II (TβRII) TGF-β receptors are transmembrane serine–threonine kinases while the type III receptor (TβRIII), a membrane bound proteoglycan also known as beta-glycan, lacks kinase activity. TβRIII, which also exists in a soluble form, can covalently bind two TGF-β molecules increasing the concentration of TGF-β at the cell surface and maximizing the interaction with type I and II receptors (Lopez-Casillas et al. 1993). TGF-β bound to TβRII recruits TβRI to execute transphosphorylation (Wrana et al. 1994). TβRI subsequently phosphorylates receptor-regulated Smad proteins 2 and 3 (R-Smads). Smad2/3 proteins can either interact with Smad4 and translocate to the nucleus to initiate gene transcription or can interact with inhibitory Smad (I-Smads) proteins (i.e Smad7) that functionally inhibit the cascade (see below). Once within the nucleus, Smad2/3/4 forms a nuclear complex with additional cell-specific co-transcription factors. These complexes bind to and regulate the transcription of a variety of target promoters and genes, depending on the type of co-factors present (Massague & Chen 2000). Massague and coworkers used transcriptomic profiling to show that there are groups of genes that seem to be commonly regulated after TGF-β stimulation of three different human epithelial cell lines (HaCat skin keratinocytes, MCF-10A breast epithelial cells and HPL1 lung epithelial cells) (Kang et al. 2003). TGF-β stimulation regulated the expression of over 100 genes in at least two of these cell lines. Functional grouping of these genes demonstrated the broad spectrum of TGF-β effects on epithelial cell function. Genes were clustered in groups regulating cytostatic program, extracellular matrix, paracrine network, signaling network, transcriptional network, negative feedback and other responses (Kang et al. 2003).
TGF-β signaling and proteasomal activity
Mechanisms for feedback regulation of TGF-β signaling are beginning to be elucidated. Members of the I-Smad subclass, Smad6 and Smad7, bind activated TβRI, prevent phosphorylation of R-Smads and thus their nuclear translocation, and recruit E3-type ubiquitin ligases to the receptors complexes, ultimately leading to their degradation (Ebisawa et al. 2001, Derynck & Zhang 2003, Shi & Massague 2003). For instance, the E3 ligase Smad ubiquitination regulatory factor (Smurf1) targets Smads1, -4 and -5 for degradation depending on the presence of I-Smads (Zhu et al. 1999, Moren et al. 2005). Smad2 is targeted by additional E3 ligases for poly-ubiquitination and degradation (Fukuchi et al. 2001, Kuratomi et al. 2005). In contrast, Smad4 can be stabilized through mono-ubiquitination and sumoylation with consequent enhancement or inhibition of TGF-β signaling (Long et al. 2003, Ohshima & Shimotohno 2003, Liang et al. 2004). Recent evidence suggests that Smad-independent induction of E3 ligases, such as Smurf2, can act as negative feedback for TGF-β-mediated signaling (Ohashi et al. 2005). Another E3 ligase, AIP4, also inhibits TGF-β signaling (Lallemand et al. 2005). Although AIP4 specifically targets Smad7, the mechanism for AIP4-mediated TGF-β inhibition is presumably through stabilization of the Smad7/TβRI complex revealing an alternate mechanism through which ubiquitination can regulate TGF-β signaling (Lallemand et al. 2005). The preceding results demonstrate the strict interconnection between TGF-β-mediated signaling and proteasomal activity.
TGF-β regulation of cell cycle and epithelial growth control in hormone-dependent tissues
One major target of TGF-β signaling is cell cycle progression, specifically the G1–S transition. Progress through the cell cycle is controlled by two families of proteins: the cyclins and the cyclin-dependent kinases (cdks). Transitions from early to late G1 phases and late G1 to S phases require the activation of cyclin D/cdk4 and 6 and cyclin E/ cdk2 complexes respectively (Yue & Mulder 2001). These substrates are regulated by TGF-β through two major mechanisms (Dupont et al. 2004): (1) by transcriptional activation of the cyclin-dependent kinase inhibitors p21 (WAF1) and p15 (INK4b) (Seoane 2004, Seoane et al. 2002) and (2) by inhibition of the growth promoting transcription factors c-MYC and Id 1-3 (Chen et al. 2002, Kang et al. 2003, Kowanetz et al. 2004). Additionally, TGF-β regulates p53 activation (Ewan et al. 2002b). Active p53 induces p21, which inhibits the cyclin E/cdk2 complex and, together with p15, the cyclin D/cdk4 complex. Inhibition of cyclin D/cdk4 complexes prevents the hyperphosphorylation of retinoblastoma (Rb) protein and thereby G1–S phase transition, resulting in a G1 arrest.
We have examined the growth suppressive effect(s) of TGF-β at the tissue level in the mammary gland. In the normal mammary gland, TGF-β activation is restricted to the luminal epithelium. Immunofluorescence studies in tissue sections demonstrated differential activation of TGF-β within cellular subpopulations during phases of hormonal stimulation at estrus and pregnancy (Ewan et al. 2002a). Depletion of TGF-β, as measured in mice engineered to lack a copy of the Tgfβ1 gene, results in accelerated morphogenesis during puberty and increased epithelial proliferation during estrus and pregnancy. TGF-β1 has also been implicated in the proliferative response of breast cancer cells to steroid hormones (Wakefield et al. 1991). Depletion of TGF-β alone, without the influence of steroid hormones (e.g. after ovariectomy), was not sufficient to increase proliferation, suggesting a role for TGF-β in inhibiting the proliferation of steroid-sensitive cells during phases of hormonal stimulation.
Further characterization of this subpopulation of cells indicated a primary role for TGF-β1 in the estrogen response. Approximately 35% of cells showed TGF-β1 activation at estrus and co-localized with nuclear localization of Smad2/3, indicating autocrine action. Furthermore, nuclear Smad2/3 colocalized with nuclear estrogen receptor α (ERα) (Fig. 2⇓). In contrast to the uterus, mammary ERα-positive cells rarely co-localize with markers of proliferation (i.e. Ki67) in either human (Clarke et al. 1997) or rodent mammary gland (Russo et al. 1999). To determine whether TGF-β1 is responsible for the quiescence of the ERα-positive population, we examined mouse mammary epithelial glands at estrus. Decreasing gene dose of TGF-β (i.e. TGF-β heterozygous) significantly increased ERα co-localization with markers of proliferation (i.e. Ki-67 or 5-bromo-2-deoxyuridine (BrdU)) at estrus. Conversely, mammary epithelial expression of constitutively active TGF-β1, via the mouse mammary tumor virus (MMTV) promoter, suppressed proliferation of ERα-positive cells (Ewan et al. 2005).
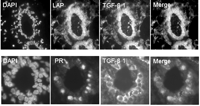
Dual immunofluorescence of TGF-β activation and hormone receptor expression at estrus in the mouse mammary gland; nuclei are counter-stained with DAPI. (Upper panel) Tissue sections of mouse mammary gland at estrus are stained with antibodies specifically recognizing latent TGF-β (LAP) and active TGF-β1. TGF-β activation at estrus is restricted to a subset of epithelial cells (merged image). (Lower panel) Dual staining with an antibody recognizing progesterone receptor (PR) reveals that TGF-β activation is restricted to the PR/ER-positive cell population during estrus (merged image).
These results suggest a possible explanation for the changes in ERα frequency within human mammary breast and their relationship to breast cancer risk. Several authors have suggested that misregulation of the ERα proliferative population may contribute to the genesis of breast cancer (Shoker et al. 1999, Frech et al. 2005). Furthermore, the frequency of ERα cells in human breast increase with age and other factors that correlated with breast cancer risk (Khan et al. 1994, Lawson et al. 1999, 2002). Thus, if TGF-β regulation of the ERα subpopulation is a key component of mammary homeostasis, dysregulation of TGF-β could expand the ERα subpopulation. Taken together, these results demonstrate that TGF-β1 activation functionally restrains ERα-positive cells from proliferating in the adult mammary gland. In a recent paper Wu et al. (2003) identified Smad4 as an ERα co-repressor providing a mechanism for the crosstalk between TGF-β and estrogen. We propose that TGF-β deregulation during ageing may promote the proliferation of ERα-positive cells associated with breast cancer risk in humans.
A different example for the interplay between TGF-β and steroid hormone effects can be found in the prostate. Androgens are required in the prostate epithelium to promote growth and development (Danielpour 2005). Androgen withdrawal on the other hand leads to a dramatic apoptotic cell death (English et al. 1987) accompanied by an up-regulation of TGF-β ligands, receptors and the activation of Smads in the involuting tissue (Kyprianou & Isaacs 1989, Kyprianou et al. 1991, Kim et al. 1996). This apoptotic response can be provoked in rats by implanting TGF-β pellets into the prostate gland (Martikainen et al. 1990). In homeostasis there is a delicate balance between the growth promoting effects of androgens and the apoptotic effects of TGF-β. During human prostate carcinogenesis epithelial cells develop a resistance to TGF-β-mediated growth inhibition, which is paralleled by a down-regulation of TβRI and II (Guo et al. 1997). TGF-β exerts its effects not only directly on the epithelial compartment but also indirectly through mediating the interaction of prostate epithelial cells and the surrounding stroma.
The physiological role of TGF-β is less well characterized in the human endometrium, which undergoes cyclic proliferation under the influence of steroid hormones. The expression of TGF-β in the endometrium through the menstrual and estrous cycles and pregnancy has been investigated in only a few studies. However, there is no sufficient information available on the cell-specific and temporal activation pattern of the TGF-β isoforms (Godkin & Dore 1998). As in other tissues, TGF-β seems to be involved in growth regulation and at least the reported expression levels in endometrial epithelium and stroma show an estrous/menstrual cycle-dependent and therefore hormone-dependent pattern (Gold et al. 1994, Marshburn et al. 1994).
TGF-β and tamoxifen
Tamoxifen (Tam) is a selective estrogen receptor modulator (SERM) and the current standard adjuvant treatment for ER-positive breast cancers. By inhibiting the binding of estrogen to its receptor, Tam has been shown to improve disease-free and overall survival of patients with initial positive hormone receptor status. Tam exercises its effects by inhibiting proliferation and increasing apoptosis. A major drawback in Tam treatment is that a considerable number of patients relapse due to the development of Tam resistance. The exact mechanisms are poorly understood, but over-expression of TGF-β has been implicated in this phenomenon (Thompson et al. 1991).
The administration of Tam itself has been shown to produce increased systemic and local levels of TGF-β. Using an MCF-7 ER-positive breast cancer cell line, Chen and colleagues showed that incubation with Tam inhibited cellular growth, induced apoptosis, up-regulated TGF-β mRNA and activated TGF-β (Chen et al. 1996). Moreover, inhibition of TGF-β2, using antisense oligonucleotides, restored Tam sensitivity in an antiestrogen resistant human breast cancer cell (LCC2) (Arteaga et al. 1999). Thus, Tam induced over-expression of TGF-βs, and a constitutional over-expression of TGF-β by the progressing tumor can synergistically contribute to Tam resistance. On the other hand, the growth inhibitory effect of Tam is not completely abrogated after transfection of a dominant negative TβRII into MCF-7 cells, showing that TGF-β is not the exclusive mediator of Tam action (Koli et al. 1997).
TGF-β in mammary development
TGF-β is involved in embryogenesis, establishment of the embryonic axis, inducing meso- and endoderm, patterning the nervous system and determining the left/right asymmetry in vertebrates (for overview see Schier 2003). The murine mammary gland development is an excellent model for TGF-β-mediated regulation of epithelial growth and differentiation (for review see Barcellos-Hoff & Ewan 2000). Under the hormonal effects of puberty (from 3–8 weeks of age in mice) the mammary tree is established in the mouse mammary fat pad. A multicellular functional unit, termed the ‘end bud’, is present at the tip of every developing duct. An outer layer of so-called cap cells invades the fat pad whereas cells in the center of the end bud apoptose forming a luminal structure. Once ductal development is complete, repeated estrous cycles elicit further elaboration, countered by minor involution, of the epithelium generating small lateral branches. The upsurge of hormones during pregnancy induces a massive lobuloalveolar differentiation of the epithelium increasing the epithelial cell mass from about 10% in the gland of nulliparous animals to as much as 90% in the gland of pregnant animals. Upon weaning, involution destroys the majority of the secretory units allowing the gland repeated cycles of growth and differentiation.
The contributions of TGF-β to these processes have been investigated through experimentation with exogenous TGF-β stimulation or pre-incubation with TGF-β neutralizing antibodies, as well as within transgenic mice with manipulated levels of TGF-β. Daniel and colleagues showed that exogenous administration of TGF-β during puberty leads to a reversible regression of end buds (Daniel et al. 1989). Interestingly, a similar protocol during pregnancy-induced growth did not impede alveolar morphogenesis (Daniel et al. 1989). If constitutively active TGF-β is expressed under the MMTV-promoter, the gland is transiently hypoplastic during ductal morphogenesis but recovers and is able to undergo full lactational differentiation (Pierce et al. 1993). If constitutively active TGF-β is expressed under the whey acidic protein (WAP)-promoter, a milk protein expressed during pregnancy and lactation, alveolar development is compromised but ductal morphogenesis is unaffected (Jhappan et al. 1993). These two mouse models, using developmentally restricted promoters, illustrate that TGF-β inhibits proliferation in response to either the hormones of puberty or pregnancy. Tgfβ1 heterozygote mice, in which TGF-β levels are reduced by 90%, show accelerated ductal outgrowth during puberty and alveolar expansion during pregnancy but have a grossly normal phenotype in the adult gland (Ewan et al. 2002a). The frequency of proliferating epithelial cells is significantly higher in Tgfβ1 heterozygote mice than in wild-type mice, as also occurs in other epithelial organs such as the liver (Böttinger et al. 1997), but appears to be compensated for by increased apoptosis.
Cheng and colleagues have conditionally knocked out TβRII selectively in mouse fibroblasts (Cheng et al. 2005). Interestingly, the mice showed a significant phenotype at 6 weeks of age with reduced ductal elongation and end bud size. Thirty percent of animals exhibited mammary gland tissue devoid of mature ducts and terminal end buds. This study showed that a loss of TGF-β signaling in the stroma altered paracrine signaling to the mammary epithelium (Cheng et al. 2005) and thereby impaired normal mammary gland development.
TGF-β and mammary stem cells
Transplantation experiments performed by DeOme in the 1950s were the first evidence for the existence of tissue-specific mammary epithelial stem cells (DeOme et al. 1959). These studies demonstrated that a complete and functional mammary gland could be regenerated by transplantation of small fragments from virtually every part of the donor gland. As established in hematopoietic stem cell research, the regeneration of a functional organ after transplantation has been the gold standard for demonstrating the existence of cells with stem cell self-renewal and differentiation capacity. The biology of the mouse mammary gland provides the opportunity to perform transplantation experiments without the interference of the host epithelium since the mammary gland tree develops from a glandular rudiment that can be removed before puberty and growth begins around 3 weeks of age. This results in a gland free, referred to as a ‘cleared’, mammary fat pad (CFP) suitable for receiving the donor tissue at the time of clearing or later. The transplantation of mammary fragments results in outgrowths that resemble the normal mammary gland morphology and function. Using the CFP technique, Sakakura (1983) showed that the morphogenesis by epithelial cells is a function of the supporting stroma. When salivary epithelium is transplanted to the mammary stroma, it exhibits the simple ductal branching pattern of mammary epithelium; conversely, mammary epithelium combined with salivary mesenchyme generates the complex pattern typical of salivary gland.
A number of studies have been conducted to determine the effects of TGF-β on mammary epithelial stem cell function (Robinson et al. 1991, Kordon et al. 1995, Boulanger & Smith 2001, Buggiano et al. 2001). Consistent with its general growth inhibitory function, ectopic expression of constitutively active TGF-β1 under the WAP promoter was shown to lead to premature senescence of the mammary stem cell population as shown by a decrease in serial transplantation capacity and failure of the gland to transform into a lactating phenotype (Boulanger & Smith 2001). The potential for TGF-β-mediated premature aging of mammary stem cells led to investigations into the putative therapeutic benefits of targeting tumor stem cell-like compartments with TGF-β. Boulanger & Smith (2001) injected the breast cancer inducing MMTV in both wild-type mice and those over expressing constitutively active TGF-β under the WAP promoter. Only 1 of 17 animals in the TGF-β group compared with 15 of 29 wild-type animals developed tumors in the 18 months after injection. These results infer a positive correlation between the lifespan of the mammary stem cell and cancer risk and a supervisory and inhibitory role for TGF-β over both. The extent to which TGF-β influences mammary stem cell functions beyond the inhibition of proliferation (which is not specific to stem cells) has so far not been investigated but is an interesting subject for future investigations.
A recent publication from Wilson’s group provides further insight into the regulating effects of TGF-β on stem cells. Prostatic stem cells are located in the mostly quiescent proximal region of the prostate gland (Tsujimura et al. 2002). Cells in this region also frequently over-express BCL-2, which protects them from apoptosis. Immunostaining for latent and active TGF-β showed that TGF-β activation was differential along the different parts of the prostate gland (Salm et al. 2005). In homeostasis, the cells in the proximal ‘stem cell’ region produced and activated significantly more TGF-β than cells in the distal part. Androgen withdrawal resulted in an increase in distal TGF-β activation, which led to apoptosis of cells in this region. At the same time, the proximal cells decreased TGF-β signaling allowing stem cells to proliferate in response to growth factors. In addition, proximal cells were more resistant to the differentiation inducing effects of TGF-β than the remaining cells (Salm et al. 2005).
Recently, the implications of stem cells for tumorigenesis have invigorated investigations into this theoretical cellular compartment (Reya et al. 2001). The line of argument is that a stem cell might be a target for carcinogenesis because it is long-lived, can easily accumulate damage, and might be able to conserve damage because of its slow cyclic pattern. The group of Max Wicha and Michael Clarke recently showed that a restricted subset of human breast cancer cells, defined by a combination of surface markers, has the ability to generate tumors in nude mice (Al-Hajj et al. 2003). As these cells show the generally postulated stem cell features of self-renewal and production of phenotypically heterogeneous progeny, they concluded that such cells might be considered tumor stem cells. However, the identification of their possible origin from tissue-specific stem cells has not been made due to the lack of suitable markers for the identification of normal epithelial stem cells.
TGF-β and its dual role in cancer
Due to its growth suppressive and apoptotic effects within many non-transformed epithelial lines, TGF-β is considered a tumor suppressor during the initial stages of carcinogenesis (Roberts & Wakefield 2003, Siegel & Massague 2003). The physiological activities of TGF-β in mediating growth regulation, DNA damage responses, apoptosis, as well as the maintenance of tissue integrity and chromosomal stability may explain aspects of its tumor suppressive roles (Glick et al. 1996) (Fig. 3A⇓). At later stages of carcinogenesis, however, TGF-β promotes tumor progression through the induction of epithelial to mesenchymal transition (EMT) (Roberts & Wakefield 2003, Siegel & Massague 2003) (Fig. 3B⇓). The following section will outline the dual nature of TGF-β, and its interplay with hormonal effects, within tumorigenesis. Emphasis will be placed upon the role of TGF-β within radiation-induced mammary carcinogenesis. As ionizing radiation (IR) represents a well-established carcinogen (Gofman 1990) and IR induces stress responses at the cellular (i.e. p53 activation and growth arrest), tissue (i.e. apoptosis) and endocrine level, IR-induced mammary carcinogenesis is an exceptional model to examine the duality of TGF-β in hormonally regulated cancers.
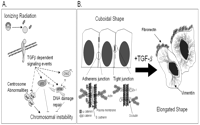
Dual role of TGF-β in carcinogenesis. TGF-β signaling has been shown to function within both tumor suppression and tumor promotion. (A) In addition to its well-described role regulating G1 transition, TGF-β facilitates cellular responses to DNA damage, including the activation of p53 and the suppression of cells with irradiation-induced centrosomal amplification. Potential targets to facilitate TGF-β-mediated p53 activation include ATM and Chk2. Thus, TGF-β signaling is essential for the maintenance of genetic stability within mammary epithelia. (B) At later stages, transformed cells resist the growth suppressive effects of TGF-β signaling. Many reports demonstrate that TGF-β signaling promotes the loss of epithelial characteristics, such as adherins and tight junctions with down-regulation of E-cadherin and Zo-1 respectively, and the acquisition of mesenchymal characteristics, such as increased intracellular vimentin and secretion of fibronectin. These TGF-β-mediated transitions require, or are accompanied by, concurrent activation of multiple growth-promoting pathways including the MAPK and phosphatidylinositol 3-kinase (PI3K).
TGF-β functions as a tumor suppressor through p53 activation, growth arrest and apoptosis
The seminal importance of hormones to mammary tumorigenesis is highlighted by epidemiological data that demonstrates a 50% reduction in breast cancer risk subsequent to one full term pregnancy (Rosner et al. 1994). It was postulated that the protective effect of hormones in mammary carcinogenesis might, in part, be related to regulation of p53 activation (Becker et al. 2005). Proof of principle experiments examining p53-deficient mammary tissues demonstrated a lack of parity-induced protection, confirming the participation of p53 in hormonal-mediated chemoprevention (Medina & Kittrell 2003). The tumor suppressor p53 is the major sensor and signal to promote apoptosis and cell cycle arrest following cellular stress. The apoptotic response eliminates cells with carcinogenic potential whereas cell cycle arrest provides time to accomplish DNA repair. The p53-mediated cellular stress response includes fast, posttranslational modifications of p53 protein stability following DNA damage. The changes include stabilization and tetramerization of the p53 protein through a complex series of covalent p53 protein modifications such as serine phosphorylation and de-phosphorylation. Once activated, p53 acts at the trailhead of transcriptional, cell cycle, repair and apoptotic responses.
Radiation-induced TGF-β activation and signaling, like p53 stability, is rapid (Ewan et al. 2002b). Moreover, TGF-β gene status significantly impacts cellular damage response. Radiation-induced apoptosis is absent in Tgfβ1 +/− mammary epithelium and cell cycle arrest is a function of TGF-β gene status in embryo epithelium (Ewan et al. 2002b). TGF-β signaling does not affect the abundance of p53 protein but rather its posttranslational modification and stabilization (Ewan et al. 2002b). Recent analyses demonstrate a direct cross talk between TGF-β and hormones in the process of p53 activation within irradiated mammary epithelium. In ovariectomized mice, systemic injections of estrogen and progesterone were necessary to recover maximal expression of cell cycle regulators following ionizing radiation (Becker et al. 2005). While ovarian steroid hormone administration augmented the p53 response to radiation (Becker et al. 2005), neutralization of TGF-β blocked responsiveness (Ewan et al. 2002b). The surprising conclusion from these experiments is that TGF-β, representing an extracellular signaling molecule, determines p53 response to DNA damage caused by radiation.
TGF-β and p53 display many similarities and some events originally attributed to p53 are actually directly inducible by TGF-β: GADD-45 and WAF/p21 can be induced by TGF-β treatment of transformed keratinocytes without functional p53 (Landesman et al. 1997) and TGF-β activates c-jun amino terminal kinase involved in UV-mediated apoptosis (Merryman et al. 1998). On the other hand, p53 action largely overlaps with TGF-β effects (Cordenonsi et al. 2003, Takebayashi-Suzuki et al. 2003). Mutant p53 correlates with reduced TGF-β responsiveness in human bronchial epithelial cells (Gerwin et al. 1992), murine keratinocytes (Reiss et al. 1993) and thyroid epithelial cells (Wyllie et al. 1991). In the early phase of mammary involution, TGF-β leads to an eightfold increase in p21/WAF mRNA, and p53 can be detected on transcriptional as well as on protein level (Strange et al. 1992, Jerry et al. 1998). However, the radiation-induced p21 response is absent in p53 −/−mice (Strange et al. 1992). Interestingly, p53 participates in TGF-β signaling (Cordenonsi et al. 2003), which complicates interpretation but suggests a mutual enhancement of response to damage.
The mechanisms by which ovarian steroid hormones and TGF-β act to increase the p53 response in the mammary epithelium are of potential therapeutic interest. Jerry and coworkers ruled out direct DNA damage as well as direct transcriptional regulation as potential mechanisms by which these hormones regulate p53 activation (Becker et al. 2005). Moreover, as TGF-β gene dose affects radiation-induced p53 phosphorylation and not total protein, it is likely that estrogen + progesterone and TGF-β regulate p53 through an indirect posttranslational mechanism (Ewan et al. 2002b). Such a mechanism could incorporate additional binding factors, such as chk2 (checkpoint kinase 2), ATM (ataxia telangiectasia mutated), plk1 (polo-like kinase 1) and BRCA1, that affect p53 activation (Chehab et al. 1999, Khosravi et al. 1999, Somasundaram et al. 1999, Hirao et al. 2002 Ando et al. 2004). The relative abundance of these gene products may be altered subsequent to TGF-β or hormonal signaling with consequent alterations in p53 stability. Alternatively, TGF-β and hormones may directly alter p53 stability. Mechanistically the ERα complexes with p53 protein, leading to stabilization of p53 (Liu et al. 2000) and altered transcriptional responses to estrogen (Liu et al. 1999). TGF-β signaling, through the induction of Smads, may alter hormonal receptor responses. Some ERα-positive breast cancer cells are refractory to TGF-β-mediated growth arrest due to the reduced expression of TβRI and TβRII through transcriptional repression (Kim et al. 2000). Smads are induced by antiestrogens (Buck et al. 2004), have been shown to bind ERα (Matsuda et al. 2001, Yamamoto et al. 2002, Wu et al. 2003) and may represent the point of cross talk between these two signaling pathways. Indeed, activation of a bone morphogenetic protein (BMP)/Smad1 pathway characterizes breast cancers and is a major hallmark of the progression and dedifferentiation of estrogen-positive breast cancer (Helms et al. 2005). Thus, steroid hormones and TGF-β likely cooperate to balance the induction of factors that alter the activation and stabilization of p53, leading to elimination of damaged and potentially oncogenic cells.
TGF-β regulation of genomic stability through the centrosome and proteasome
p53 has been defined as a quintessential tumor suppressor through its ability to regulate apoptosis, DNA repair and cell cycle arrest (reviewed in Brown & Attardi 2005, Sengupta & Harris 2005). However, recent examination of p53 function and localization illustrate an intimate association between p53 and a small sub-cellular organelle named the centrosome (reviewed in Tarapore & Fukasawa 2002). The centrosome, named for its frequent localization at the cell center, is the major microtubule organizer in many cell types. In addition to maintaining cytoskeletal architecture through nucleation and organization of microtubules, the centrosome participates in a number of processes including cell motility, polarity, signaling, damage responses and division. During cell division, the centrosome is duplicated once during S phase and it is these duplicated centrosomes that functionally nucleate the bipolar mitotic apparatus necessary for division. p53 regulates centrosomal fidelity and loss of p53 induces centrosomal abnormalities with consequent genetic instability (reviewed in Tarapore & Fukasawa 2002, Salisbury et al. 2004). Importantly, centrosomal abnormalities, defined based upon abnormal number, size or shape, have been identified in the majority of tumors examined (Pihan et al. 1998) and have been implicated in the generation of chromosomal instability (CIN) and tumorigenesis (Pihan et al. 2001, 2003, Salisbury et al. 2004).
In primary human breast cancer, centrosomal abnormalities are prevalent, occur early (Salisbury et al. 2004) and are significantly associated with hormone receptor status (Schneeweiss et al. 2003). However, progesterone increases aneuploidy in p53 null tumor cells without associated centrosomal amplification (Goepfert et al. 2000). In a rodent model of estrogen-mediated mammary tumorigenesis, estrogen supplementation induced over-expression of a regulatory centrosomal protein, Aurora A kinase, as well as near complete prevalence of centrosomal amplification in mammary tumors accompanied with genetic aberrations (Li et al. 2004). While there are strong interconnections between hormonal exposure, centrosomal amplification and mammary tumorigenesis, to date there are no published direct links between exogenous TGF-β and centrosomal dysregulation.
Glick and colleagues have shown that Tgfβ1 null keratinocytes demonstrate elevated genomic instability, as measured by N-(phosphonacetyl)-l-aspartic acid (PALA)-induced gene amplification, and lack the typical PALA-induced, p53-dependent growth arrest despite functionally wild-type p53 activity (Glick et al. 1996). Moreover, v-rasHa transduced Tgfβ1 null keratinocytes rapidly developed aneuploidy with multiple mitotic aberrations (Glick et al. 1996, 1999). Given the intimate association between TGF-β, p53 activation and aneuploidy, we investigated whether TGF-β, like p53, also participates in the regulation of centrosomal amplification. Compromised TGF-β production or signaling in either murine keratinocytes or human mammary epithelial cells induces elevated centrosomal amplification in spite of functional p53 within these non-malignant epithelial cells (authors’ unpublished observations). Centrosomal amplification subsequent to irradiation has been reported in cancer cell lines of various species and is almost invariably associated with a prolonged G2 cell cycle arrest (Sato et al. 1983, 2000, Shono et al. 2001, Dodson et al. 2004, Kawamura et al. 2004, Yoon et al. 2005). Interestingly, the absence of exogenous TGF-β sensitizes irradiated, p53-competent human mammary epithelial cells (HMEC) to persistent centrosomal amplification and the addition of TGF-β impairs the permanence, but not induction, of these abnormalities (authors’ unpublished observations). These results suggest that p53-competent HMEC require TGF-β signaling to properly supervise irradiation-induced cellular stress.
An additional consideration for these TGF-β-mediated processes may be the regulation of the proteasome. While it is beyond the scope of this review, it is warranted to mention that irradiation dramatically changes the composition of the proteasome as well as its activity, with subsequent consequences on DNA repair, cell cycle progression (through regulation of cyclin E, p53 and p21 expression) and cell death (reviewed in McBride et al. 2003). The proteasome is also intimately associated with centrosomal stability, as various components of the proteasome/ubiquitin pathway localize to centrosomes (Freed et al. 1999, Wigley et al. 1999, Nakayama et al. 2000) and inhibition of proteasomal activity is sufficient to induce multipolar spindle phenotypes (Ehrhardt & Sluder 2005). As outlined above, the role of the proteasome/ubiquitin pathway in regulating TGF-β signaling is well established. Hormones and their receptors also affect and are regulated by ubiquitin-mediated protein degradation (Hamel et al. 2004, Laios et al. 2005), while radiation rapidly inhibits proteosome function (McBride et al. 2003).
One gene product that highlights the interconnectivity of hormones, TGF-β, centrosomes and proteasomal activity is breast cancer 1 (BRCA1). The BRCA1 gene was first localized by genetic linkage in 1994 and loss of function mutations of BRCA1 have been reported to confer up to an 82% risk of developing breast cancer and a 54% risk of developing ovarian cancer by the age of 80 years (reviewed in Kennedy et al. 2004). The major role of BRCA1 is to respond to DNA damage and thus it is important in DNA repair, transcriptional regulation, cell cycle regulation, proteolysis and centrosomal stability (Kennedy et al. 2004). BRCA1 localizes to centrosomes during mitosis (Hsu & White 1998), interacts directly with the key centrosomal structural protein γ-tubulin (Hsu et al. 2001) and may serve as a negative regulator of centrosomal duplication (reviewed in Deng 2002). Indeed, disruption of BRCA1 function induces centrosomal amplification specifically within mammary cell lines (Starita et al. 2004). Mechanistically, BRCA1 and its binding partner BARD1, together a highly active E3 ubiquitin ligase (Hashizume et al. 2001), target γ-tubulin turnover through mono-ubiquitination; loss of this regulation, through inhibition of BRCA1 or alteration of γ-tubulin, results in mammary-specific centrosome amplification (Starita et al. 2004). Reduction of BRCA1 also disrupts mammary epithelial cell morphogenesis within an in vitro 3D culture system in a manner that is reversible by addition of unidentified soluble factors (Furuta et al. 2005).
While hereditary breast cancers, associated with germ-line BRCA1 mutations, are not associated with a higher frequency of TβR inactivation than sporadic cases (Xie et al. 2002), increasing evidence suggests significant cross-talk between TGF-β signaling and BRCA1 function. TGF-β1 inhibits BRCA1 expression in a Rb-dependent manner within Mv1Lu cells (Satterwhite et al. 2000). Swift, an important constituent of embryonic TGFβ-induced gene transcription, contains a BRCA C-terminal (BRCT) domain that directly interacts with and co-activates Smad2 (Shimizu et al. 2001). Smad3 also directly interacts with the BRCT domain of BRCA1 and TGF-β/Smad3-modified BRCA1-dependent repair of DNA double strand (Dubrovska et al. 2005). TGF-β-mediated regulation of BRCA1, and other DNA damage response proteins such as p53 and ATM, may dramatically alter centrosomal amplification, proteasomal activity and genetic stability following irradiation.
The preceding studies demonstrating TGF-β-mediated supervision of centrosomal abnormalities may indicate reciprocity between TGF-β and hormonal signaling, proteasomal activity and centrosomal stability. Given these intimate associations, it is likely that cellular homeostasis is maintained through the cumulative cross talk of extracellular signals and intracellular regulatory mechanisms. Carcinogens such as ionizing radiation may disrupt this homeostasis and result in intracellular aberrations (i.e. centrosomal abnormalities, increased/aberrant proteasomal activity) that are supervised by extracellular cues. To some extent, TGF-β mediates the proper supervision of these aberrations. However, this supervision may be incomplete in some contexts such as in early preneoplastic lesions in which TGF-β signaling is perturbed or lost. Within the context of incomplete supervision and subsequent persistent aberrations, TGF-β signaling can cooperate with ionizing radiation to promote cellular programs that are tumorigenic, such as EMT.
TGF-β during tumor progression and EMT
Several lines of evidence underscore the transition of TGF-β as it switches from tumor suppression to tumor promotion. Paradoxically for a tumor suppressor, when compared with normal tissue, production of TGF-β is increased in breast (Travers et al. 1988, Gorsch et al. 1992, Dalal et al. 1993), gastric (Maehara et al. 1999) and prostate cancer (Steiner et al. 1994). Inhibition of TGF-β reduces tumor cell motility and metastasis in genetically induced murine mammary tumors (Muraoka et al. 2002) and over-expression of active TGF-β in vivo accelerates metastases of transgenic mammary tumors (Muraoka-Cook et al. 2004). In addition to increased TGF-β production, expression levels of I-Smads are often altered in tumor microenvironments. Smad7 is expressed at very low levels in epithelial tissues, but it is up-regulated in human pancreatic (Kleeff et al. 1999), endometrial (Dowdy et al. 2005) and colorectal cancers (Korchynskyi et al. 1999). While Smad7 can induce tumorigenicity by blocking growth arrest and apoptosis within a TGF-β sensitive cell line (Halder et al. 2005), Smad7 over-expression can also functionally inactivate Rb without interfering with Smad2/3 nuclear translocation (Boyer & Korc 2005). Thus, these cells are competent for TGF-β modulation of gene expression, such as increased plasminogen activator inhibitor (PAI)-1 expression in response to TGF-β, but are resistant to the growth inhibitory mechanisms of TGF-β signaling (Boyer & Korc 2005). These results have implication for cancer cells that are responsive to TGF-β-mediated EMT but unresponsive to cell cycle inhibition and apoptosis.
Many tumor-promoting aspects of TGF-β are related to tumor cell motility and metastasis. One mechanism by which TGF-β can promote motility is through the induction of epithelial to mesenchymal transition (EMT) (Akhurst & Balmain 1999). Cells undergoing EMT must counteract TGF-β-mediated growth control, dramatically alter shape and dissolve tight junctions and down-regulate epithelial markers and remodel cytoskeletal networks to acquire motile phenotypes. Cumulatively, these transformations can facilitate increased migration and metastasis (Thiery 2002). However, induction of EMT by TGF-β alone is a rare event in vitro (Brown et al. 2004). The rarity of TGF-β-mediated EMT in non-malignant cells is likely attributable to the requirement for simultaneous activation of multiple signaling pathways orchestrating proliferation, survival and differentiation.
TGF-β role in overcoming growth restrictions during EMT
Several reports suggest that an independent stimulus for cell proliferation must synergistically proceed or at least accompany the induction of EMT (Gotzmann et al. 2004). Consequently, constitutive activation of downstream signaling pathways, such as MAP kinase (MAPK) or PI3K, may be a requirement to induce a pre-malignant state and endow epithelial cells with an increased rate of proliferation (Gotzmann et al. 2004). In addition to counteracting the anti-proliferative and apoptotic effects of TGF-β, these cells must functionally switch differentiation programs. Recent evidence indicates a central role for Disabled-2 (Dab-2) within some of these processes. During the promotion of EMT within non-transformed murine mammary gland cells, TGF-β induces the expression of Dab-2 (Prunier & Howe 2005), a protein that functions in the regulation of the cytoskeleton and epithelial differentiation (Sheng et al. 2000). TGF-β also induces Dab-2 accumulation at the membrane and consequent binding to β1-integrin with subsequent integrin activation and protection from apoptosis (Prunier & Howe 2005). Another important intermediate is integrin-linked kinase (ILK). ILK expression is responsive to TGF-β treatment and ectopic expression of ILK induces E-cadherin and fibronectin expression and assembly within renal tubular epithelial cells (Li et al. 2003). Moreover, kinase dead ILK largely abolishes EMT within renal epithelia (Li et al. 2003). Similar results have been demonstrated within human keratinocytes; within these cells, ILK is required for TGF-β-mediated EMT through propagation of the PI3K–AKT pathway (Lee et al. 2004). These results underscore the importance of coincident activation of pathways that promote cell survival, in this case integrin-dependent cell adhesion and survival, during TGF-β-induced EMT.
TGF-β role in dissolution of tight junctions during EMT
Many signaling pathways are important for the acquisition of EMT including the canonical Ras pathway, PI3K and the MAPK pathways (Grunert et al. 2003, Thiery 2003). In addition to Ras pathways, TGF-β signaling cooperates with β1 integrin, PI3K, RhoA and protein kinase (PK)C-ζ to induce cell scattering, defined as loss of polarity and epithelial markers combined with gain of a mesenchymal gene program and migratory phenotypes (Grunert et al. 2003). Recent high throughput protein–protein interactome mapping of the TGF-β pathway has identified novel, potential EMT targets including key regulators of epithelial cytoskeletal networks, polarity and structural components of tight junctions (Barrios-Rodiles et al. 2005, Ozdamar et al. 2005). One such component of tight junctions, occludin (OCLN), interacts with TβRII in a TGF-β-dependent manner and dominant negative OCLN prevents TGF-β-dependent dissolution of tight junctions during EMT (Barrios-Rodiles et al. 2005). Recent investigations within tubular cells demonstrate the importance of cell–cell junctions and their disruption during tissue injury and repair in the initiation of EMT (Masszi et al. 2004). Indeed, the authors suggested a two-hit model during which both an initial injury, leading to losses of cell–cell contacts and redistribution of β-catenin, and TGF-β are required for EMT (Masszi et al. 2004). In normal murine mammary gland cells, another key participant in TGF-β-dependent dissolution of tight junctions is Par6 (Ozdamar et al. 2005). Par6, like OCLN, interacts with TGF-β receptors and Par6 is phosphorylated by TβRII leading to an association with Smurf1 and the localized degradation of RhoA (Ozdamar et al. 2005). Par6 mutants, inhibition of proteasomal activity or mutation of RhoA acceptor sites for ubiquitin block TGF-β-induced tight junction dissolution (Ozdamar et al. 2005). Interestingly, morphological changes similar to EMT (reduced OCLN, tight junctions and E-cadherin) were observed in estrogen receptor β knockout mice (Forster et al. 2002) and exogenous 17β-estradiol modulates occludin expression probably through post translational stabilization (Zeng et al. 2004). Moreover, Par6 was identified as a target gene of steroid receptor coactivator-3 (SRC-3) (Labhart et al. 2005), a co-activator of ERα-dependent gene regulation (Suen et al. 1998). As with p53 activation, cross-talk between hormonal and TGF-β-mediated signaling pathways may be a vital determinant of EMT-induced structural changes.
The effects of TGF-β inhibitors within cancer models
An understanding of the multiple and complex mechanisms of TGF-β regulation of epithelial function, and ultimate loss of function, will be critical in the design of novel therapeutic interventions for endocrine-related cancers. The restricted and stringently regulated activation of TGF-β found in normal tissue contrasts with the elevated TGF-β expression observed in tumors.
Mouse models of early stage epithelial cancers suggest that most are sensitive to TGF-β growth inhibition (Akhurst 2002). Early stage cancers may be suppressed by TGF-β-mediated alterations in the stroma composition and stimulation of immune system surveillance (Akhurst 2002). Older women with a TGFβ1 polymorphism, which results in more TGF-β secretion, are less likely to develop breast cancer (Ziv et al. 2001), and as mentioned before, one of the most effective breast cancer prevention therapeutics, tamoxifen, appears to induce TGF-β. As would be expected, tumors routinely develop traits that circumvent TGF-β inhibition, yet surprisingly continue to produce and may even increase their ability to activate TGF-β. Essentially, all studies to date indicate that TGF-β is increased in tumors versus normal tissue. TGF-β immunoreactivity correlates with breast cancer progression (Gorsch et al. 1992), abnormal stroma (McCune et al. 1992), and metastases (Dalal et al. 1993). Human tumors also exhibit elevated TGF-β mRNA (Barrett-Lee et al. 1990, Murray et al. 1993), immunoreactivity (Butta et al. 1992, Dublin et al. 1993, Mahara et al. 1994) and protein (Godden et al. 1993). Thus, TGF-β has been targeted for pharmacological manipulation in cancer diagnosis and therapy (Dickens & Colletta 1993, Yingling et al. 2004). But the broad range and complex timing of events that are potentially modulated by TGF-β is a challenge for pharmaceutical exploitation.
Several strategies have arisen from studies of the basic biology of TGF-β. Thrombospondin was shown to bind and activate LTGF-β (Schultz-Cherry et al. 1994). Recent structural studies have identified a thrombospondin RKPK peptide sequence that can both block re-association of LAP and TGF-β1, thereby neutralizing the ability of TGF-β to bind its receptors, and induce activation, presumably by disrupting LAP-mature TGF-β interactions (Young & Murphy-Ullrich 2004). Decorin, an extracellular proteoglycan, can also inhibit TGF-β (Border et al. 1992, Kolb et al. 2001). These naturally occurring inhibitors of TGF-β are presumably part of an environmental control to prevent rampant TGF-β activation, and may provide insight into the selectivity of its effects in vivo.
Recent studies have examined small molecule inhibitors of TGF-β. A novel small molecule of quinazoline-derived inhibitors of the type I transforming growth factor receptor was shown to be effective in cell culture. This molecule inhibits the kinase by binding to the ATP-binding site to keep the kinase in its inactive conformation (Ge et al. 2004). Another member (SB-505124) of a new class of small molecule inhibitors related to imidazole inhibitors of p38 inhibits the TGF-β-type I receptor serine/threonine kinase known as activin receptor-like kinase (ALK) 5. Selectively and concentration dependently this compound inhibits ALK 5-, and also ALK 4 (activin receptor)- and ALK 7 (nodal receptor)-dependent activation of downstream cytoplasmic signal transducers, Smad2 and Smad3 and of TGF-β-induced MAPK pathway components (DaCosta et al. 2004). Interestingly, it is selective in that it was shown not to alter ALK 1-, 2-, 3-, or 6-induced Smad signaling. Since integrins can also mediate activation of LTGF-β, the ability of a small-molecule inhibitor of integrin αvβ3, SB-223245, to block the interaction may be a means of blocking TGF-β (Ludbrook et al. 2003).
Neutralizing antibodies to TGF-β have also been used in animal models. Anti-TGF-β monoclonal antibodies prevent the cyclosporine-induced increase in the number of metastases (Hojo et al. 1999), block radiation-induced collagen remodeling (Ehrhart et al. 1997) and Smad signaling (Schultze-Mosgau et al. 2004), and inhibit establishment of MDA-231 tumors and lung metastases in athymic mice (Arteaga et al. 1993).
Therapeutic potential of inhibitors of TGF-β
Considering the pleiotropic effects of TGF-β, the timing and duration of inhibition is likely to be critical to the ultimate benefit. There may be scenarios in which TGF-β inhibition in conjunction with cancer therapy is beneficial. The most developed experimental evidence is that from radiotherapy, where there is a clear benefit in experimental normal tissue toxicity models for blocking TGF-β. Russo and colleagues have used a small molecular weight molecule, halofuginone, to block TGF-β signaling using a connective tissue radiation damage model (Xavier et al. 2004). Halofuginone inhibits the TGF-β signaling pathway by elevating inhibitory Smad7, blocking formation of phospho-Smad2 and phospho-Smad3 and decreasing cytosolic and membrane TGF-β type II receptor. Halofuginone treatment significantly lessened radiation-induced fibrosis but showed neither general toxicity nor interference with tumor control by radiation in mice. Interestingly, it appears that even transient inhibition is beneficial, which is likely to be rooted in the self-amplification of autocrine loops by TGF-β and its mediators. Transient expression of soluble TGF-β type II receptor by adenovirus infection shows promise in some experimental models of normal lung damage following radiotherapy (Rabbani et al. 2003a, Nishioka et al. 2004). It is likely that interruption of TGF-β signaling significantly affects its own production, thus limiting the source of protein as well as the means to activate it. Studies in a rat lung radiation-injury model by Vujaskovic and colleagues have shown that blocking TGF-β via expression of soluble type II receptor decreases reactive oxygen species (ROS), and blocking ROS by pharmaceuticals or by over-expression of superoxide dismutase decreases TGF-β (Rabbani et al. 2003a,b), consistent with a detrimental cascade in which TGF-β is activated by ROS, which in turn stimulates ROS, that can then activate more TGF-β. Even transient interruption of this process has significant long-term benefit by limiting normal tissue damage.
Summary
The 25-year history of TGF-β has been productive and informative as to fundamental growth control by extracellular factors and the loss of such regulation during cancer progression. TGF-β has confounded, perplexed and rewarded researchers who have struggled to understand its complex biology. The relatively recent appreciation of its role in modulating response to hormones offers a new perspective on its potential application to the treatment of endocrine-related cancers.
Funding
This work was funded, in part, by the Office of Health and Environmental Research, Health Effects Division, United States Department of Energy (contract no.03-76SF00098) and the National Institute of Environmental Health Sciences, NIH, grant number U01 ES012801 to M-H B-H. The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Footnotes
-
↵* (M C Fleisch and C A Maxwell contributed equally to this work)
- © 2006 Society for Endocrinology
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
The pleiotropic roles of TGF-β on homeostasis, disease and carcinogenesis within a variety of endocrine organs. Effects have been described in, but are not necessarily restricted to, the attributed organs.










